
Akutt lymfoblastisk leukemi - Acute lymphoblastic leukemia
| Akutt lymfoblastisk leukemi | |
|---|---|
| Andre navn | Akutt lymfatisk leukemi, akutt lymfoid leukemi |
 | |
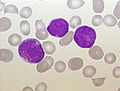
| Benmargsaspirat fra en person med forløper B-celle ALL. De store lilla cellene er lymfoblaster. | |
| Spesialitet | Hematologi , onkologi |
| Symptomer | Trøtthet, blek farge, feber, lett blødning eller blåmerker, bein smerter, forstørrede lymfeknuter |
| Komplikasjoner | Infeksjon , tumorlysesyndrom |
| Vanlig start | 2–5 år gammel |
| Typer | B-celle ALL , T-celle ALL |
| Årsaker | Vanligvis ukjent |
| Risikofaktorer | Enegget tvilling med ALL, Down syndrom , Fanconi anemi , ataksi telangiectasia , Klinefelter syndrom , høy fødselsvekt , betydelig stråling eksponering |
| Diagnostisk metode | Blodprøver og benmargsundersøkelse |
| Differensialdiagnose | Smittsom mononukleose , akutt myeloid leukemi , lymfoblastisk lymfom , aplastisk anemi |
| Behandling | Kjemoterapi , stamcelletransplantasjon , strålebehandling , målrettet terapi |
| Prognose |
Barn : 90% overlevelse fem år Voksne : 35% femårig overlevelse |
| Frekvens | 1 av 1750 barn |
| Dødsfall | 111 000 (2015) |
Akutt lymfoblastisk leukemi ( ALL ) er en kreft i lymfoidlinjen av blodceller preget av utvikling av et stort antall umodne lymfocytter . Symptomene kan være trøtthet, blek hudfarge, feber , lett blødning eller blåmerker, forstørrede lymfeknuter eller beinpine. Som en akutt leukemi utvikler ALL seg raskt og er vanligvis dødelig i løpet av uker eller måneder hvis den ikke behandles.
I de fleste tilfeller er årsaken ukjent. Genetiske risikofaktorer kan omfatte Downs syndrom , Li-Fraumeni syndrom eller nevrofibromatose type 1 . Miljømessige risikofaktorer kan omfatte betydelig stråleeksponering eller tidligere cellegift . Bevis angående elektromagnetiske felt eller plantevernmidler er uklare. Noen antar at en unormal immunrespons på en vanlig infeksjon kan være en utløser. Den underliggende mekanismen involverer flere genetiske mutasjoner som resulterer i rask celledeling . De overdrevne umodne lymfocytter i beinmargen forstyrrer produksjonen av nye røde blodlegemer , hvite blodlegemer og blodplater . Diagnosen er vanligvis basert på blodprøver og benmargsundersøkelse .
ALL behandles vanligvis først med cellegift som tar sikte på remisjon . Dette blir deretter fulgt av ytterligere cellegift vanligvis over en årrekke. Behandlingen inkluderer vanligvis også intratekal kjemoterapi siden systemisk kjemoterapi kan ha begrenset penetrasjon i sentralnervesystemet og sentralnervesystemet er et vanlig sted for tilbakefall av akutt lymfoblastisk leukemi.
Behandlingen kan også omfatte strålebehandling hvis spredning til hjernen har skjedd. Stamcelletransplantasjon kan brukes hvis sykdommen gjentar seg etter standard behandling. Ytterligere behandlinger som kimær antigenreseptor T -celle immunterapi blir brukt og studert videre.
ALLE påvirket rundt 876 000 mennesker globalt i 2015 og resulterte i rundt 111 000 dødsfall. Det forekommer oftest hos barn, spesielt de mellom to og fem år. I USA er det den vanligste årsaken til kreft og død av kreft blant barn. ALL er kjent for å være den første spredte kreften som ble kurert. Overlevelse for barn økte fra under 10% på 1960 -tallet til 90% i 2015. Overlevelsesraten er fortsatt lavere for babyer (50%) og voksne (35%). Ifølge National Cancer Intelligence Network (NCIN), generelt for mennesker med ALLE: rundt 70 av 100 mennesker (70%) vil overleve leukemien i 5 år eller mer etter at de er diagnostisert.
Tegn og symptomer
De første symptomene kan være uspesifikke, spesielt hos barn. Over 50%av barna med leukemi hadde en eller flere av fem funksjoner: en lever man kan føle (64%), en milt man kan føle (61%), blek hudfarge (54%), feber (53%) og blåmerker (52%). I tillegg kan tilbakevendende infeksjoner, trøtthet, smerter i armer eller ben og forstørrede lymfeknuter være fremtredende trekk. De B-symptomer , slik som feber, nattsvette, og vekttap, er ofte til stede i tillegg.
Symptomer på sentralnervesystemet (CNS) som kraniale nevropatier på grunn av meningeal infiltrasjon er identifisert hos mindre enn 10% av voksne og mindre enn 5% av barn, spesielt modne B-celle ALL (Burkitt leukemi) ved presentasjon.
Tegn og symptomer på ALL er varierende og inkluderer:
- Generalisert svakhet og trøtthet
- Anemi
- Svimmelhet
- Hodepine, oppkast, sløvhet, nakkestivhet eller kranialnervelammelse (CNS -involvering)
- Hyppig eller uforklarlig feber og infeksjon
- Vekttap og/eller tap av matlyst
- Overdreven og uforklarlig blåmerker
- Beinsmerter, leddsmerter (forårsaket av spredning av "blast" -celler til beinoverflaten eller inn i leddet fra marghulen)
- Pustløshet
- Forstørrede lymfeknuter, lever og/eller milt
- Pitting ødem (hevelse) i underekstremitetene og/eller magen
- Petekkier, som er bitte små røde flekker eller streker i huden på grunn av lave blodplatenivå
- Testikkelforstørrelse
- Mediastinal masse
Årsaken

Kreftcellen i ALL er lymfoblasten. Normale lymfoblaster utvikler seg til modne, infeksjonskampende B-celler eller T-celler, også kalt lymfocytter . Signaler i kroppen styrer antall lymfocytter, så verken for få eller for mange blir laget. I ALLE blir både normal utvikling av noen lymfocytter og kontrollen over antall lymfoide celler defekte.
ALT dukker opp når en enkelt lymfoblast får mange mutasjoner til gener som påvirker utvikling av blodceller og spredning. I barndommen ALL begynner denne prosessen ved unnfangelsen med arv av noen av disse genene. Disse genene øker igjen risikoen for at flere mutasjoner vil oppstå i utvikling av lymfoide celler. Enkelte genetiske syndromer, som Downs syndrom , har samme effekt. Miljørisikofaktorer er også nødvendig for å skape nok genetiske mutasjoner til å forårsake sykdom. Bevis for miljøets rolle sees i barndommen ALL blant tvillinger, der bare 10–15% av begge genetisk identiske tvillingene får ALT. Siden de har de samme genene, forklarer forskjellige miljøeksponeringer hvorfor en tvilling får ALT og den andre ikke.
Infant ALL er en sjelden variant som forekommer hos babyer under ett år. KMT2A (tidligere MLL ) genarrangementer er vanligst og forekommer i embryoet eller fosteret før fødselen. Disse omorganiseringene resulterer i økt ekspresjon av blodcelleutviklingsgener ved å fremme gentranskripsjon og gjennom epigenetiske endringer. I motsetning til ALL i barndommen antas ikke miljøfaktorer å spille en vesentlig rolle. Bortsett fra KMT2A -omorganiseringen , finnes vanligvis bare en ekstra mutasjon. Miljøeksponering er ikke nødvendig for å skape flere mutasjoner.
Risikofaktorer
Genetikk
Vanlige arvelige risikofaktorer inkluderer mutasjoner i ARID5B , CDKN2A / 2B , CEBPE , IKZF1 , GATA3 , PIP4K2A og, mer sjelden, TP53 . Disse genene spiller viktige roller i mobilutvikling, spredning og differensiering. Individuelt er de fleste av disse mutasjonene lav risiko for ALLE. Betydelig risiko for sykdom oppstår når en person arver flere av disse mutasjonene sammen.
Ujevn fordeling av genetiske risikofaktorer kan bidra til å forklare forskjeller i sykdomsrater blant etniske grupper. For eksempel er ARID5B -mutasjonen mindre vanlig i etniske afrikanske populasjoner.
Flere genetiske syndrom har også økt risiko for ALL. Disse inkluderer: Downs syndrom , Fanconi anemi , Bloom syndrom , X-bundet agammaglobulinemi , alvorlig kombinert immunsvikt , Shwachman-Diamond syndrom , Kostmann syndrom , neurofibromatose type 1 , ataksi-telangiectasia , paroksysmal nattlig hemoglobinuri og Li-Fraumeni syndrom . Færre enn 5% av tilfellene er forbundet med et kjent genetisk syndrom.
Sjeldne mutasjoner i ETV6 og PAX5 er forbundet med en familiær form for ALLE med autosomal dominant mønster av arv .
Miljø
Miljøeksponeringene som bidrar til fremveksten av ALL er omstridt og gjenstand for pågående debatt.
Høye nivåer av stråleeksponering fra kjernefysisk nedfall er en kjent risikofaktor for å utvikle leukemi. Bevis på om mindre stråling, som fra røntgenbilder under graviditet, øker risikoen for sykdom, er fortsatt avgjørende. Studier som har identifisert en sammenheng mellom røntgenbilder under graviditet og ALL fant bare en litt økt risiko. Eksponering for sterk elektromagnetisk stråling fra kraftledninger har også vært assosiert med en litt økt risiko for ALL. Dette resultatet stilles spørsmålstegn ved at det ikke er kjent noen årsaksmekanisme som forbinder elektromagnetisk stråling med kreft.
Høy fødselsvekt (større enn 4000 g eller 8,8 kg) er også forbundet med en liten økt risiko. Mekanismen som forbinder høy fødselsvekt med ALL er heller ikke kjent.
Bevis tyder på at sekundær leukemi kan utvikle seg hos personer som behandles med visse typer cellegift, for eksempel epipodofyllotoksiner og cyklofosfamid .
Infeksjoner
Det er noen bevis på at en vanlig infeksjon, som influensa , indirekte kan fremme fremveksten av ALL. Forsinket infeksjon-hypotesen sier at ALLE skyldes et unormalt immunrespons på infeksjon hos en person med genetiske risikofaktorer. Forsinket utvikling av immunsystemet på grunn av begrenset eksponering for sykdom kan resultere i overdreven produksjon av lymfocytter og økt mutasjonshastighet under en sykdom. Flere studier har identifisert lavere forekomst av ALLE blant barn med større eksponering for sykdom tidlig i livet. Svært små barn som går i barnehagen har lavere priser på ALLE. Bevis fra mange andre studier som ser på eksponering av sykdom og ALL, er ikke avgjørende. Noen forskere har knyttet hygienehypotesen .
Mekanisme
Flere karakteristiske genetiske endringer fører til dannelsen av en leukemisk lymfoblast. Disse endringene inkluderer kromosomale translokasjoner , intrakromosomale omorganiseringer , endringer i antall kromosomer i leukemiske celler og ytterligere mutasjoner i individuelle gener. Kromosomale translokasjoner innebærer å flytte et stort område av DNA fra ett kromosom til et annet. Dette trekket kan resultere i å plassere et gen fra ett kromosom som fremmer celledeling til et mer aktivt transkribert område på et annet kromosom. Resultatet er en celle som deler seg oftere. Et eksempel på dette omfatter den translokasjon av C-MYC , et gen som koder for en transkripsjonsfaktor som fører til økt celledeling, ved siden av immunoglobulin tung - eller lettkjede gen- forsterkere , som fører til økt C-MYC -ekspresjon og øket celledelingen . Andre store endringer i kromosomstruktur kan resultere i plassering av to gener rett ved siden av hverandre. Resultatet er kombinasjonen av to vanligvis separate proteiner til et nytt fusjonsprotein . Dette proteinet kan ha en ny funksjon som fremmer utviklingen av kreft. Eksempler på dette inkluderer ETV6 - RUNX1 -fusjonsgenet som kombinerer to faktorer som fremmer blodcelleutvikling og BCR - ABL1 -fusjonsgenet til Philadelphia -kromosomet . BCR - ABL1 koder for en alltid aktivert tyrosinkinase som forårsaker hyppig celledeling. Disse mutasjonene produserer en celle som deler seg oftere, selv i fravær av vekstfaktorer .
Andre genetiske endringer i B-celle ALL inkluderer endringer i antall kromosomer i de leukemiske cellene. Å få minst fem ekstra kromosomer, kalt høy hyperdiploidi, forekommer oftere. Mindre ofte går kromosomer tapt, kalt hypodiploidy , som er assosiert med en dårligere prognose. Ytterligere vanlige genetiske endringer i B-celle ALL involverer ikke-arvelige mutasjoner til PAX5 og IKZF1 . I T-celle kan ALL, LYL1 , TAL1 , TLX1 og TLX3 omorganiseres.
ALLE resultater når nok av disse genetiske endringene er tilstede i en enkelt lymfoblast. I barndommen ALL, for eksempel, blir en fusjonsgen-translokasjon ofte funnet sammen med seks til åtte andre ALL-relaterte genetiske endringer. Den første leukemiske lymfoblasten kopierer seg selv til et stort antall nye lymfoblaster, hvorav ingen kan utvikle seg til fungerende lymfocytter. Disse lymfoblastene bygger seg opp i benmargen og kan spre seg til andre steder i kroppen, for eksempel lymfeknuter , mediastinum , milten , testiklene og hjernen , noe som fører til vanlige symptomer på sykdommen.
Diagnose
Diagnostisering av ALL begynner med en grundig sykehistorie, fysisk undersøkelse , fullstendig blodtelling og blodflekker. Selv om mange symptomer på ALLE finnes ved vanlige sykdommer, gir vedvarende eller uforklarlige symptomer mistanke om kreft. Fordi mange funksjoner i sykehistorien og eksamen ikke er spesifikke for ALLE, er det ofte nødvendig med ytterligere testing. Et stort antall hvite blodlegemer og lymfoblaster i sirkulerende blod kan være mistenkelige for ALLE fordi de indikerer en rask produksjon av lymfoide celler i marg. Jo høyere disse tallene vanligvis tyder på en dårligere prognose. Selv om antall hvite blodlegemer ved første presentasjon kan variere betydelig, sees sirkulerende lymfoblastceller på perifere blodutstryk i de fleste tilfeller.
En benmargsbiopsi gir avgjørende bevis på ALLE, vanligvis med> 20% av alle cellene som er leukemiske lymfoblaster. En lumbal punktering (også kjent som en ryggmarg) kan avgjøre om ryggraden og hjernen er invadert. Hjerne- og ryggsøyle -involvering kan diagnostiseres enten gjennom bekreftelse av leukemiske celler i lumbalpunksjonen eller gjennom kliniske tegn på CNS -leukemi som beskrevet ovenfor. Laboratorietester som kan vise abnormiteter inkluderer blodtelling, nyrefunksjon, elektrolytt og leverenzymtester.
Patologisk undersøkelse, cytogenetikk (spesielt tilstedeværelsen av Philadelphia -kromosom ) og immunofenotyping fastslår om de leukemiske cellene er myeloblastiske (nøytrofile, eosinofiler eller basofiler) eller lymfoblastiske ( B -lymfocytter eller T -lymfocytter ). Cytogenetisk testing på margprøvene kan bidra til å klassifisere sykdom og forutsi hvor aggressivt sykdomsforløpet vil være. Ulike mutasjoner har vært assosiert med kortere eller lengre overlevelse. Immunhistokjemisk testing kan avsløre TdT- eller CALLA -antigener på overflaten av leukemiske celler. TdT er et protein uttrykt tidlig i utviklingen av pre-T og pre-B celler, mens CALLA er et antigen som finnes i 80% av ALLE tilfeller og også i "blast crisis" av CML .
Medisinsk bildebehandling (for eksempel ultralyd eller CT -skanning ) kan finne invasjon av andre organer, vanligvis lunge , lever, milt, lymfeknuter, hjerne, nyrer og reproduktive organer.
akutt lymfoblastisk leukemi (ALL), perifert blod av et barn, Pappenheim flekk, forstørrelse x100

beinmargsutstryk (stor forstørrelse) fra en person med akutt lymfoblastisk leukemi

beinmargsmerke fra en person med akutt lymfoblastisk leukemi



Immunofenotyping
I tillegg til cellemorfologi og cytogenetikk, er immunofenotyping , en laboratorieteknikk som brukes til å identifisere proteiner som uttrykkes på celleoverflaten, en sentral komponent i diagnosen ALL. Den foretrukne metoden for immunfenotyping er gjennom flytcytometri . I maligne lymfoblaster av ALL kan uttrykk for terminal deoksynukleotidyltransferase (TdT) på celleoverflaten bidra til å differensiere maligne lymfocyttceller fra reaktive lymfocytter , hvite blodlegemer som reagerer normalt på en infeksjon i kroppen. På den annen side uttrykkes myeloperoksidase (MPO), en markør for myeloidavstamning , vanligvis ikke. Fordi forløper B -celle og forløper T -celler ser like ut, kan immunofenotyping bidra til å differensiere undertypen ALL og modenhetsnivået til de ondartede hvite blodcellene. Undertyper av ALL bestemt av immunofenotype og i henhold til modningstrinnene.
| B -celle Lineage | T -celle Lineage |
|---|---|
| pre-pre-B ALL (pro-B-ALL) | forløper T- ALL |
| felles ALL | moden T-celle ALL |
| pre-B ALL | |
| moden B -celle ALL (Burkitt leukemi - FAB L3) |
Et omfattende panel av monoklonale antistoffer mot celleoverflatemarkører, spesielt CD eller klynge av differensieringsmarkører, brukes til å klassifisere celler etter slekt. Nedenfor er immunologiske markører assosiert med B -celle og T -celle ALL.
| Immunologiske markører | B -celle Lineage | T -celle Lineage |
|---|---|---|
| B -celle Lineage | ||
| CD19, CD22, CD79a | + | - |
| CD10 | - eller + (vanlig ALL) | |
| cytoplasmatisk Ig | - eller + (pre-B ALL) | |
| overflate Ig | - eller + (moden B-celle ALL) | |
| TdT | + | + |
| T -celle Lineage | ||
| CD2, CD3, CD4, CD5, CD7, CD8 | - | + |
| TdT | + | + |
Cytogenetikk
Cytogenetisk analyse har vist forskjellige proporsjoner og frekvenser av genetiske abnormiteter i tilfeller av ALLE fra forskjellige aldersgrupper. Denne informasjonen er spesielt verdifull for klassifisering og kan delvis forklare de forskjellige prognosene for disse gruppene. Når det gjelder genetisk analyse, kan tilfeller stratifiseres i henhold til ploidi , et antall sett med kromosomer i cellen og spesifikke genetiske abnormiteter, for eksempel translokasjoner . Hyperdiploide celler er definert som celler med mer enn 50 kromosomer, mens hypodiploid er definert som celler med mindre enn 44 kromosomer. Hyperdiploid -tilfeller har en tendens til å ha en god prognose, mens hypodiploid -tilfeller ikke gjør det. For eksempel er den vanligste spesifikke abnormiteten i barndommen B -ALL t (12; 21) ETV6 - RUNX1 -translokasjon, der RUNX1 -genet, som koder for et protein involvert i transkripsjonell kontroll av hemopoiesis , har blitt translokert og undertrykt av ETV6 - RUNX1 fusjonsprotein.
Nedenfor er en tabell med frekvensene til noen cytogenetiske translokasjoner og molekylære genetiske abnormiteter i ALLE.
| Cytogenetisk translokasjon | Molekylær genetisk abnormitet | % |
|---|---|---|
| kryptisk t (12; 21) | TEL - AML1 fusjon | 25,4% |
| t (1; 19) (q23; p13) | E2A - PBX ( PBX1 ) fusjon | 4,8% |
| t (9; 22) (q34; q11) | BCR-ABL fusjon (P185) | 1,6% |
| t (4; 11) (q21; q23) | MLL - AF4 fusjon | 1,6% |
| t (8; 14) (q24; q32) | IGH - MYC -fusjon | |
| t (11; 14) (p13; q11) | TCR - RBTN2 fusjon |
Klassifisering
Fransk-amerikansk-britisk
Historisk sett, før 2008, ble ALL klassifisert morfologisk ved hjelp av det fransk-amerikansk-britiske (FAB) systemet som i stor grad var avhengig av morfologisk vurdering. FAB-systemet tar hensyn til informasjon om størrelse, cytoplasma , nukleoli , basofili (farge på cytoplasma) og vakuolering ( boblelignende egenskaper).
| FAB -undertype | Cell Type | Kjennetegn | Kommentarer |
|---|---|---|---|
| ALLE - L1 | T-celle eller pre-B-celle | Små og homogene (ensartede) celler | |
| ALL - L2 | T-celle eller pre-B-celle | Store og heterogene (varierte) celler | |
| ALL - L3 | B -celle | Store og varierte celler med vakuoler | Eldre B-celle ALL heter også Burkitt leukemi. Vanligvis dårlig prognose med standardterapi |
Selv om noen klinikere fortsatt bruker FAB -ordningen for å beskrive tumorcellens utseende, har mye av denne klassifiseringen blitt forlatt på grunn av den begrensede virkningen på behandlingsvalg og prognostisk verdi.
Verdens Helseorganisasjon
I 2008 ble Verdens helseorganisasjons klassifisering av akutt lymfoblastisk leukemi utviklet i et forsøk på å lage et klassifiseringssystem som var mer klinisk relevant og kunne gi meningsfylte prognostiske og behandlingsbeslutninger. Dette systemet gjenkjente forskjeller i genetiske, immunofenotype , molekylære og morfologiske trekk som ble funnet gjennom cytogenetiske og molekylære diagnostiske tester. Denne undertypingen hjelper til med å bestemme prognosen og den mest hensiktsmessige behandlingen for hvert enkelt tilfelle av ALL.
WHO -undertyper knyttet til ALLE er:
- B-lymfoblastisk leukemi/lymfom
- Ikke annet spesifisert (NOS)
- med tilbakevendende genetiske abnormiteter
- med t (9; 22) (q34.1; q11.2); BCR-ABL1
- med t (v; 11q23.3); KMT2A omorganisert
- med t (12; 21) (p13.2; q22.1); ETV6-RUNX1
- med t (5; 14) (q31.1; q32.3) IL3-IGH
- med t (1; 19) (q23; p13.3); TCF3-PBX1
- med hyperdiploidi
- med hypodiploidy
- T-lymfoblastisk leukemi/lymfom
- Akutte leukemier av tvetydig avstamning
- Akutt udifferensiert leukemi
- Blandet fenotype akutt leukemi (MPAL) med t (9; 22) (q34.1; q11.2); BCR-ABL1
- MPAL med t (v; 11q23.3); KMT2A omorganisert
- MPAL, B/myeloid, NOS
- MPAL, T/myeloid, NOS
Behandling

Målet med behandlingen er å indusere en varig remisjon , definert som fravær av påvisbare kreftceller i kroppen (vanligvis mindre enn 5% blastceller i beinmargen).
I løpet av de siste tiårene har det vært fremskritt for å øke effekten av behandlingsregimer, noe som resulterer i økte overlevelsesrater. Mulige behandlinger for akutt leukemi inkluderer cellegift , steroider , strålebehandling , intensive kombinerte behandlinger (inkludert benmarg eller stamcelletransplantasjoner ), målrettet terapi og/eller vekstfaktorer.
Kjemoterapi
Kjemoterapi er den første behandlingen du velger, og de fleste med ALLE får en kombinasjon av medisiner. Det er ingen kirurgiske alternativer på grunn av kroppsfordelingen av de ondartede cellene . Generelt kombinerer cytotoksisk kjemoterapi for ALL flere antileukemiske legemidler som er skreddersydd for hver person. Kjemoterapi for ALL består av tre faser: remisjon induksjon, intensivering og vedlikeholdsterapi.
| Fase | Beskrivelse | Agenter |
|---|---|---|
| Remisjon induksjon | Tar sikte på å:
Må overvåke nøye for tumorlysesyndrom etter at behandling er startet Overvåkning av første respons på behandlingen er viktig ettersom manglende evne til å vise clearance av blod eller benmargsblast i løpet av de første 2 ukene av behandlingen har vært forbundet med en høyere risiko for tilbakefall
Start profylakse på CNS og administrer intratekal kjemoterapi via Ommaya reservoar eller flere lumbale punkteringer |
Kombinasjon av:
Profylakse i sentralnervesystemet kan oppnås via:
I Philadelphia kromosom -positive ALL kan intensiteten av den første induksjonsbehandlingen være mindre enn det tradisjonelt er gitt. |
| Konsolidering/intensivering | Bruk høye doser cellegift for å redusere tumorbyrden ytterligere | Typiske protokoller bruker følgende gitte blokker (varierer fra 1-3 blokker avhengig av personens risikokategori) i forskjellige kombinasjoner av flere medikamenter:
Tilbakefall i sentralnervesystemet behandles med intratekal administrering av hydrokortison , metotreksat og cytarabin. |
| Vedlikeholdsterapi | Drep alle restceller som ikke ble drept av remissionsinduksjon og intensiveringsregimer
|
Typisk protokoll vil omfatte:
|
På grunn av tilstedeværelsen av CNS -involvering hos 10–40% av voksne med ALL ved diagnosen, starter de fleste tilbydere profylakse og behandling av sentralnervesystemet (CNS) under induksjonsfasen, og fortsetter den under konsoliderings-/intensiveringsperioden.
Voksen cellegiftbehandling etterligner de i barndommen ALL; er imidlertid knyttet til en høyere risiko for tilbakefall av sykdom bare med cellegift. Det bør være kjent at 2 undertyper av ALL (B-celle ALL og T-celle ALL) krever spesielle hensyn når det gjelder valg av et passende behandlingsregime hos voksne med ALL. B-celle ALL er ofte assosiert med cytogenetiske abnormiteter (spesifikt t (8; 14), t (2; 8) og t (8; 22)), som krever aggressiv terapi som består av korte, høyintensive regimer. T-celle ALL reagerer mest på cyklofosfamidholdige midler.
Siden kjemoterapiregimene kan være intensive og langvarige, har mange mennesker et intravenøst kateter satt inn i en stor vene (betegnet et sentralt venekateter eller en Hickman -linje ), eller en Portacath , vanligvis plassert i nærheten av kragebenet, for lavere infeksjonsrisiko og langsiktig levedyktighet av enheten. Hanner tåler vanligvis et lengre behandlingsforløp enn kvinner, ettersom testiklene kan fungere som et reservoar for kreft.
Strålebehandling
Strålebehandling (eller strålebehandling) brukes på smertefulle benete områder, ved høye sykdomsbyrder eller som en del av forberedelsene til en beinmargstransplantasjon (total kroppsbestråling). Tidligere brukte leger vanligvis stråling i form av helhjernestråling for profylakse i sentralnervesystemet, for å forhindre forekomst og/eller tilbakefall av leukemi i hjernen. Nyere studier viste at CNS kjemoterapi ga resultater som gunstige, men med færre utviklingsbivirkninger. Som et resultat har bruken av helhjernestråling vært mer begrenset. De fleste spesialister på leukemi hos voksne har forlatt bruken av strålebehandling for forebygging av CNS, i stedet for å bruke intratekal kjemoterapi.
Biologisk terapi
Valg av biologiske mål på grunnlag av deres kombinatoriske effekter på leukemiske lymfoblaster kan føre til kliniske studier for forbedring av effektene av ALL behandling. Tyrosinkinasehemmere (TKI), for eksempel imatinib , er ofte inkorporert i behandlingsplanen for personer med Bcr-Abl1+ (Ph+) ALL. Imidlertid er denne undertypen ALL ALL motstandsdyktig mot kombinasjonen av cellegift og TKI, og allogen stamcelletransplantasjon anbefales ofte ved tilbakefall.
Immunterapi
Kimære antigenreseptorer (CAR) er utviklet som en lovende immunterapi for ALLE. Denne teknologien bruker et enkelt kjede variabelt fragment (scFv) designet for å gjenkjenne celleoverflatemarkøren CD19 som en metode for behandling av ALL.
CD19 er et molekyl som finnes på alle B-celler og kan brukes som et middel for å skille den potensielt ondartede B-cellepopulasjonen. I denne terapien blir mus immunisert med CD19-antigenet og produserer anti-CD19-antistoffer. Hybridomer utviklet fra musemiltceller smeltet til en myelomcellelinje kan utvikles som en kilde for cDNA som koder for det CD19 -spesifikke antistoffet. CDNA blir sekvensert og sekvensen som koder for de variable tunge og variable lette kjedene til disse antistoffene klones sammen ved bruk av en liten peptidlinker . Denne resulterende sekvensen koder for scFv. Dette kan klones inn i et transgen , som koder for det som blir endodomenet til CAR. Varierende ordninger for underenheter fungerer som endodomenet, men de består generelt av hengselområdet som fester seg til scFv, en transmembranregion, den intracellulære regionen til et kostimulatorisk molekyl som CD28 , og det intracellulære domenet til CD3 -zeta som inneholder ITAM -gjentakelser . Andre sekvenser som ofte inkluderes er: 4-1bb og OX40 . Den endelige transgensekvensen, som inneholder scFv- og endodomainsekvensene, blir deretter satt inn i immuneffektorceller som er hentet fra personen og ekspandert in vitro . I forsøk har disse vært en type T-celle som er i stand til cytotoksisitet .
Innsetting av DNA i effektorcellen kan utføres ved flere metoder. Vanligvis gjøres dette ved hjelp av et lentivirus som koder for transgenet. Pseudotypede, selvinaktiverende lentivirus er en effektiv metode for stabil innsetting av et ønsket transgen i målcellen. Andre metoder inkluderer elektroporering og transfeksjon , men disse er begrenset i effektivitet ettersom transgenuttrykk avtar over tid.
De genmodifiserte effektorcellene transplanteres deretter tilbake til personen. Vanligvis utføres denne prosessen i forbindelse med et kondisjoneringsprogram som cyklofosfamid , som har vist seg å potensere effekten av infunderte T-celler. Denne effekten har blitt tilskrevet å lage et immunologisk rom der cellene befolker seg. Prosessen som helhet resulterer i en effektorcelle , typisk en T-celle, som kan gjenkjenne et tumorcelle- antigen på en måte som er uavhengig av det store histokompatibilitetskomplekset og som kan starte en cytotoksisk respons.
I 2017 ble tisagenlecleucel godkjent av FDA som en CAR-T- behandling for personer med akutt B-celle lymfoblastisk leukemi som ikke reagerte tilstrekkelig på andre behandlinger eller har fått tilbakefall. I en 22-dagers prosess er "stoffet" tilpasset hver person. T -celler renset fra hver person blir modifisert av et virus som setter inn gener som koder for en kimær antigenreseptor i deres DNA, en som gjenkjenner leukemiceller.
Tilbakefall ALLE
Vanligvis har personer som opplever tilbakefall i ALL etter første behandling en dårligere prognose enn de som forblir i fullstendig remisjon etter induksjonsterapi. Det er usannsynlig at tilbakevendende leukemi vil reagere positivt på standard kjemoterapiregime som opprinnelig ble implementert, og i stedet bør disse menneskene prøves på reinduksjonskjemoterapi etterfulgt av allogen benmargstransplantasjon . Disse personene i tilbakefall kan også få blinatumomab , ettersom det har vist seg å øke remisjonsrater og generelle overlevelsesrater, uten økte toksiske effekter.
Lav dose palliativ stråling kan også bidra til å redusere tumorbyrden i eller utenfor sentralnervesystemet og lindre noen symptomer.
Nylig har det også vært bevis og godkjennelse for bruk av dasatinib , en tyrosinkinasehemmer. Det har vist effekt hos personer med Ph1-positiv og imatinibresistent ALL, men mer forskning må gjøres på langsiktig overlevelse og tid til tilbakefall.
Bivirkninger
Kjemoterapier eller stamcelletransplantasjoner kan kreve en blodplatetransfusjon for å forhindre blødning. Videre kan pasienter som gjennomgår en stamcelletransplantasjon utvikle en transplantat-mot-vert-sykdom (GvHD). Det ble evaluert om mesenkymale stromaceller kan brukes for å forhindre GvHD. Bevisene er svært usikre på den terapeutiske effekten av mesenkymale stromaceller for å behandle transplantat-mot-vert-sykdommer etter en stamcelletransplantasjon på dødelighet av alle årsaker og fullstendig forsvinning av kroniske akutte transplantat-mot-vert-sykdommer. Mesenkymale stromaceller kan resultere i liten eller ingen forskjell i dødelighet av alle årsaker, tilbakefall av ondartet sykdom og forekomst av akutte og kroniske transplantat-mot-vert-sykdommer hvis de brukes av profylaktisk årsak.
Støttende terapi
Å legge til fysiske øvelser i standardbehandlingen for voksne pasienter med hematologiske sykdommer som ALL kan resultere i liten eller ingen forskjell i dødelighet, livskvalitet og fysisk funksjon. Disse øvelsene kan resultere i en liten reduksjon i depresjon. Videre reduserer trolig aerobe fysiske øvelser tretthet. Bevisene er svært usikre på effekten på angst og alvorlige bivirkninger.
Genterapi
Brexucabtagene autoleucel (Tecartus) ble godkjent for behandling av voksne med tilbakefall eller ildfast B-celle forløper akutt lymfoblastisk leukemi i oktober 2021.
Hver dose brexucabtagene autoleucel er en tilpasset behandling som er opprettet ved hjelp av mottakerens eget immunsystem for å bekjempe lymfom. Mottakerens T -celler , en type hvite blodceller, samles og genetisk modifiseres for å inkludere et nytt gen som letter målretting og drep av lymfomceller. Disse modifiserte T -cellene infunderes deretter tilbake til mottakeren.
Prognose
Før utviklingen av cellegiftbehandlinger og hematopoietisk stamcelletransplantasjon overlevde barn en median lengde på 3 måneder, hovedsakelig på grunn av enten infeksjon eller blødning. Siden kjemoterapien kom, har prognosen for leukemi hos barn forbedret seg sterkt, og barn med ALL anslås å ha en 95% sannsynlighet for å oppnå en vellykket remisjon etter 4 ukers behandling. Personer i barneomsorg med ALL i utviklede land har en overlevelsesrate på mer enn 80% på fem år. Det anslås at 60–80% av voksne som gjennomgår induksjonskjemoterapi oppnår fullstendig remisjon etter 4 uker, og de over 70 år har en kur på 5%. Hutter JJ (juni 2010). "Barndomsleukemi". Pediatrics in Review . 31 (6): 234–41. doi : 10.1542/pir.31-6-234 . PMID 20516235 .</ref>

Imidlertid er det forskjellige prognoser for ALLE blant individer avhengig av en rekke faktorer:
- Kjønn: Kvinner har en tendens til å klare seg bedre enn menn.
- Etnisitet: Kaukasiere er mer sannsynlig å utvikle akutt leukemi enn afroamerikanere , asiater eller latinamerikanere . Imidlertid har de en tendens til å ha en bedre prognose enn ikke-kaukasiere.
- Alder ved diagnose: barn 1–10 år er mest sannsynlig å utvikle ALT og bli helbredet for det. Tilfeller hos eldre mennesker er mer sannsynlig å skyldes kromosomavvik (f.eks. Philadelphia -kromosomet) som gjør behandlingen vanskeligere og prognosene dårligere. Eldre mennesker vil også sannsynligvis ha medkomorbide medisinske tilstander som gjør det enda vanskeligere å tolerere ALLE behandlinger.
- Antall hvite blodlegemer ved diagnosen større enn 30 000 (B-ALL) eller 100 000 (T-ALL) er assosiert med dårligere utfall
- Kreft som sprer seg til sentralnervesystemet ( hjerne eller ryggmarg ) har dårligere utfall.
- Morfologiske, immunologiske og genetiske undertyper
- Personens respons på første behandling og lengre tid (mer enn 4 uker) for å nå fullstendig remisjon
- Tidlig tilbakefall av ALLE
- Minimal restsykdom
- Genetiske lidelser , som Downs syndrom og andre kromosomale abnormiteter (aneuoploidi og translokasjoner)
| Faktor | Ufordelaktig | Gunstig |
|---|---|---|
| Alder | <2 eller> 10 år | 3–5 år |
| Kjønn | Hann | Hunn |
| Løp | Svart | Kaukasisk |
| Organomegali | Tilstede | Fraværende |
| Mediastinal masse | Tilstede | Fraværende |
| CVS -involvering | Tilstede | Fraværende |
| Leukocyttall | B-ALL> 30 000 mm 3 T-ALL> 100 000 mm 3 | Lav |
| Hemogblobin konsentrasjon | > 10 g/dl | <10 g/dl |
| Celletype | Ikke lymfatisk | Lymfoid |
| Celleavstamning | Pre B -celle +
T-ALL (barn) |
Tidlig Pre B -celle |
| Karyotype | Translokasjon | Hyperdiploidi |
| Svar på behandling | Langsom
> 1 uke for å fjerne blaster fra blod |
Rask
<1 uke for å fjerne blaster fra blod |
| Tid til remisjon | > 4 uker | <4 uker |
| Minimal restsykdom | Positiv etter 3 - 6 måneder | Negativ etter 1 måned (barn) eller 3 måneder (voksne) |
Cytogenetikk, studiet av karakteristiske store endringer i kromosomene i kreftceller , er en viktig prediktor for utfall. Noen cytogenetiske undertyper har en dårligere prognose enn andre. Disse inkluderer:
- Personer med t (9,22) positive-ALL (30% av voksne ALLE tilfeller) og andre Bcr-abl- omorganiserte leukemier er mer sannsynlig å ha en dårlig prognose, men overlevelsesraten kan stige ved behandling bestående av cellegift og Bcr-abl tyrosinkinasehemmere.
- En translokasjon mellom kromosomer 4 og 11 forekommer i omtrent 4% av tilfellene og er mest vanlig hos spedbarn under 12 måneder.
| Cytogenetisk endring | Risikokategori |
|---|---|
| Philadelphia kromosom | Dårlig prognose |
| t (4; 11) (q21; q23) | Dårlig prognose |
| t (8; 14) (q24.1; q32) | Dårlig prognose |
| Kompleks karyotype (mer enn fire abnormiteter) | Dårlig prognose |
| Lav hypodiploidi eller nær triploidi | Dårlig prognose |
| Sletting av kromosom 7 | Dårlig prognose |
| Trisomi 8 | Dårlig prognose |
| Høy hyperdiploidi (trisomi 4, 10, 17) | God prognose |
| del (9p) | God prognose |
- Hyperdiploidy (> 50 kromosomer) og t (12; 21) er gode prognostiske faktorer og utgjør også 50% av alle pediatriske ALLE tilfeller.
| Prognose | Cytogenetiske funn |
|---|---|
| Gunstig | Hyperdiploidy> 50; t (12; 21) |
| Mellom | Hyperdiploidy 47–50; Normal (diploid); del (6q); Omorganisering av 8q24 |
| Ufordelaktig | Hypodiploidy-nær haploidi; Nær tetraploidi; del (17p); t (9; 22); t (11q23) |
Uklassifisert ALL anses å ha en mellomliggende prognoserisiko, et sted mellom de gode og dårlige risikokategoriene.
Epidemiologi
ALLE påvirket rundt 876 000 mennesker og resulterte i 111 000 dødsfall globalt i 2015. Det forekommer hos både barn og voksne med høyeste frekvens sett mellom tre og sju år. Rundt 75% av tilfellene forekommer før 6 -årsalderen med en sekundær økning etter 40 -årsalderen. Det anslås å ramme 1 av 1500 barn.
Med hensyn til de brede aldersprofilene til de berørte, forekommer ALLE nylig hos omtrent 1,7 per 100 000 mennesker per år. ALL representerer omtrent 20% av voksne og 80% av leukemi hos barn, noe som gjør den til den vanligste kreft i barndommen. Selv om 80 til 90% av barna vil ha en langsiktig fullstendig respons med behandling, er det fortsatt den viktigste årsaken til kreftrelaterte dødsfall blant barn. 85% av tilfellene er av B-cellelinje og har like mange tilfeller hos både menn og kvinner. De resterende 15% av T-cellelinjen har en mannlig overvekt.
Globalt forekommer ALLE vanligvis oftere hos kaukasiere, latinamerikanere og latinamerikanere enn hos afrikanere. I USA er ALL mer vanlig hos barn fra kaukasisk (36 tilfeller/millioner) og latinamerikansk (41 tilfeller/millioner) avstamning sammenlignet med barn fra afrikansk (15 tilfeller/millioner) avstamning.
Svangerskap
Leukemi er sjelden assosiert med graviditet, og rammer bare 1 av 10.000 gravide. Behandlingen av leukemi hos en gravid person avhenger først og fremst av typen leukemi. Akutte leukemier krever normalt rask, aggressiv behandling, til tross for betydelig risiko for tap av graviditet og fødselsskader , spesielt hvis cellegift gis i løpet av den utviklingsfølsomme første trimesteren .
Referanser
Eksterne linker
- Akutt lymfatisk leukemi ved American Cancer Society
- Barndom ALL behandling ved National Cancer Institute
| Klassifisering | |
|---|---|
| Eksterne ressurser |