
Immun trombocytopenisk purpura - Immune thrombocytopenic purpura
| Immun trombocytopenisk purpura | |
|---|---|
| Andre navn | Idiopatisk trombocytopenisk purpura, idiopatisk immuntrombocytopeni, primær immuntrombocytopeni, idiopatisk trombocytopenisk purpura, primær immun trombocytopenisk purpura, autoimmun trombocytopenisk purpura |
 | |
| Petechiae, eller små blåmerke-lignende markeringer, kan forekomme i ITP | |
| Spesialitet | Hematologi , generell kirurgi |
Immun trombocytopenisk purpura ( ITP ), også kjent som idiopatisk trombocytopenisk purpura eller immuntrombocytopeni , er en type trombocytopenisk purpura definert som et isolert lavt antall blodplater med normalt benmarg i fravær av andre årsaker til lave blodplater. Det forårsaker et karakteristisk rødt eller lilla blåmerke-lignende utslett og en økt tendens til å blø. To distinkte kliniske syndromer manifesterer seg som en akutt tilstand hos barn og en kronisk tilstand hos voksne. Den akutte formen følger ofte etter en infeksjon og forsvinner spontant i løpet av to måneder. Kronisk immuntrombocytopeni vedvarer lenger enn seks måneder med en spesifikk årsak ukjent.
ITP er en autoimmun sykdom med antistoffer påvisbare mot flere blodplateoverflatestrukturer .
ITP diagnostiseres ved å identifisere et lavt antall blodplater på et fullstendig blodtall (en vanlig blodprøve ). Siden diagnosen avhenger av utelukkelse av andre årsaker til lavt antall blodplater, kan imidlertid ytterligere undersøkelser (for eksempel benmargsbiopsi ) være nødvendig i noen tilfeller.
I milde tilfeller kan det være nødvendig med nøye observasjon, men svært lave tellinger eller betydelig blødning kan føre til behandling med kortikosteroider , intravenøst immunglobulin , anti-D immunglobulin eller immunsuppressive medisiner . Ildfast ITP (reagerer ikke på konvensjonell behandling eller konstant tilbakefall etter splenektomi ) krever behandling for å redusere risikoen for klinisk signifikant blødning. Trombocyttransfusjon kan brukes i alvorlige tilfeller med svært lavt antall blodplater hos mennesker som blør. Noen ganger kan kroppen kompensere ved å lage unormalt store blodplater.
Tegn og symptomer
Tegn inkluderer spontan dannelse av blåmerker (purpura) og petechiae (små blåmerker), spesielt på ekstremiteter , blødning fra neseborene og/eller tannkjøttet , og menoragi (overdreven menstruasjonsblødning ), hvorav noen kan oppstå hvis antall blodplater er lavere 20 000 per ul. Et svært lavt antall (<10 000 per μl) kan resultere i spontan dannelse av hematomer (blodmasser) i munnen eller på andre slimhinner . Blødningstiden fra mindre skader eller skrubbsår er vanligvis forlenget.
Alvorlige og muligens dødelige komplikasjoner på grunn av ekstremt lave tellinger (<5000 pr. Μl) inkluderer subaraknoid eller intracerebral blødning (blødning inne i skallen eller hjernen ), lavere gastrointestinal blødning eller annen indre blødning. En ITP -pasient med ekstremt lavt antall er sårbar for indre blødninger forårsaket av sløv mageskade , slik man kan oppleve ved et motorulykke . Disse komplikasjonene er ikke sannsynlige når blodplatetallet er over 20 000 per ul.
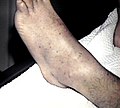
Petechiae på nedre ekstremiteter
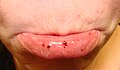
Oral petechiae/purpura - underleppe
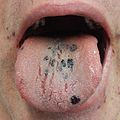
Petechia på tungen hos en person med blodplater på 3 på grunn av ITP

Petechia i underbenet hos en person med blodplater på 3 på grunn av ITP




Patogenese
I omtrent 60 prosent av tilfellene kan antistoffer mot blodplater påvises. Oftest er disse antistoffene mot blodplatemembranen glykoproteiner IIb-IIIa eller Ib-IX , og er av typen immunglobulin G (IgG). Den Harrington-Hollingsworth eksperiment etablert immun patogenesen av ITP.
Beleggingen av blodplater med IgG gjør dem mottakelige for opsonisering og fagocytose av miltmakrofager , så vel av Kupffer-celler i leveren . IgG -autoantistoffene antas også å skade megakaryocytter , forløpercellene til blodplater, selv om dette antas å bare bidra til å redusere antall blodplater. Nyere forskning tyder nå på at svekket produksjonen av glykoprotein- hormon trombopoietin , som er det middel for blodplatedannelse, kan være en medvirkende årsak til reduksjon i sirkulerende blodplater. Denne observasjonen har ført til utviklingen av en klasse av ITP-målrettede medikamenter referert til som trombopoietin reseptor agonister .
Stimuleringen for auto-antistoffproduksjon i ITP er sannsynligvis unormal T- celleaktivitet. Foreløpige funn tyder på at disse T -cellene kan påvirkes av medisiner som retter seg mot B -celler , for eksempel rituximab .
Diagnose

Diagnosen ITP er en ekskluderingsprosess. Først må det fastslås at det ikke er andre abnormiteter i blodet enn lavt antall blodplater, og ingen fysiske tegn annet enn blødning. Deretter bør sekundære årsaker (5–10 prosent av mistenkte ITP -tilfeller) utelukkes. Slike sekundære årsaker inkluderer leukemi , medisiner (f.eks kinin , heparin ), lupus erythematosus , skrumplever , HIV , hepatitt C, medfødte årsaker, antifosfolipidsyndrom , von Willebrand -faktormangel , onyalai og andre. Alle pasienter med antatt ITP bør testes for HIV og hepatitt C -virus, da antall blodplater kan korrigeres ved behandling av den underliggende sykdommen. I omtrent 2,7 til 5 prosent av tilfellene sameksisterer autoimmun hemolytisk anemi og ITP, en tilstand referert til som Evans syndrom .
Til tross for ødeleggelse av blodplater av miltmakrofager, blir milten normalt ikke forstørret. Faktisk bør en forstørret milt føre til et søk etter andre mulige årsaker til trombocytopeni. Blødningstiden er vanligvis forlenget hos ITP -pasienter. Imidlertid frarådes bruk av blødningstid i diagnosen av retningslinjene for American Society of Hematology, og en normal blødningstid utelukker ikke en blodplateforstyrrelse.
Benmargsundersøkelse kan utføres på pasienter over 60 år og de som ikke svarer på behandling, eller når diagnosen er i tvil. Ved undersøkelse av marg kan en økning i produksjonen av megakaryocytter observeres og kan hjelpe til med å etablere en diagnose av ITP. En analyse for blodplate-antistoffer er et spørsmål om klinikerens preferanse, da det er uenighet om 80 prosent spesifisiteten til denne testen er tilstrekkelig til å være klinisk nyttig.
Behandling
Med sjeldne unntak er det vanligvis ikke nødvendig å behandle basert på antall blodplater. Mange eldre anbefalinger antydet en viss terskel for blodplater (vanligvis et sted under 20,0/µl) som en indikasjon på sykehusinnleggelse eller behandling. Gjeldende retningslinjer anbefaler behandling kun i tilfeller av betydelig blødning. Behandlingsanbefalinger er noen ganger forskjellige for ITP for voksne og barn.
Steroider
Første behandling består vanligvis av administrering av kortikosteroider , en gruppe medisiner som undertrykker immunsystemet. Dosen og administrasjonsmåten bestemmes av antall blodplater og om det er aktiv blødning: i presserende situasjoner kan infusjon av deksametason eller metylprednisolon brukes, mens oral prednison eller prednisolon kan være tilstrekkelig i mindre alvorlige tilfeller. Når blodplatetallet har blitt bedre, reduseres dosen av steroid gradvis mens muligheten for tilbakefall overvåkes. 60–90 prosent vil oppleve tilbakefall under dosereduksjon eller opphør. Langsiktige steroider unngås om mulig på grunn av potensielle bivirkninger som inkluderer osteoporose , diabetes og grå stær .
Anti-D
Et annet alternativ, egnet for Rh-positive pasienter med funksjonelle milter, er intravenøs administrering av Rho (D) immunglobulin [Human; Anti-D]. Virkningsmekanismen til anti-D er ikke fullt ut forstått. Etter administrering metter imidlertid anti-D-belagte røde blodlegemekomplekser Fcγ-reseptorseter på makrofager , noe som resulterer i fortrinnsvis ødeleggelse av røde blodlegemer (RBC), og sparer derfor antistoffbelagte blodplater . Det er to anti-D-produkter indisert for bruk hos pasienter med ITP: WinRho SDF og Rhophylac. De vanligste bivirkningene er hodepine (15%), kvalme/oppkast (12%) frysninger (<2%) og feber (1%).
Steroidsparende midler
Det er økende bruk av immunsuppressiva som mykofenolatmofetil og azatioprin på grunn av deres effektivitet. I kroniske ildfaste tilfeller, hvor immun patogenesen har blitt bekreftet, er off-label bruk av vinca- alkaloid og kjemoterapimidlet vincristin kan forsøkes. Vinkristin har imidlertid betydelige bivirkninger, og bruken av den ved behandling av ITP må behandles med forsiktighet, spesielt hos barn.
Intravenøst immunglobulin
Intravenøst immunglobulin (IVIg) kan i noen tilfeller infunderes for å redusere hastigheten som makrofager bruker antistoff -merkede blodplater. Selv om det noen ganger er effektivt, er det kostbart og gir forbedringer som vanligvis varer mindre enn en måned. Likevel, når det gjelder en ITP -pasient som allerede er planlagt for operasjon som har et farlig lavt antall blodplater og har opplevd dårlig respons på andre behandlinger, kan IVIg raskt øke antall blodplater, og kan også bidra til å redusere risikoen for større blødninger ved forbigående økning trombocyttall.
Trombopoietinreseptoragonister
Trombopoietinreseptoragonister er farmasøytiske midler som stimulerer produksjon av blodplater i benmargen. I dette skiller de seg fra de tidligere diskuterte midlene som virker ved å prøve å begrense ødeleggelse av blodplater. To slike produkter er for tiden tilgjengelige:
- Romiplostim (handelsnavn Nplate) er et trombopoiesis- stimulerende Fc-peptidfusjonsprotein (peptidlegeme) som administreres ved subkutan injeksjon . Utpekt et foreldreløst legemiddel i 2003 i henhold til amerikansk lov, viste kliniske studier at romiplostim var effektivt i behandling av kronisk ITP, spesielt hos tilbakefallende pasienter etter miltomi. Romiplostim ble godkjent av United States Food and Drug Administration (FDA) for langsiktig behandling av voksen kronisk ITP 22. august 2008.
- Eltrombopag (handelsnavn Promacta i USA, Revolade i EU) er et oralt administrert middel med en effekt som ligner den på romiplostim. Det er også vist at det øker antall blodplater og reduserer blødning på en doseavhengig måte. Promacta ble utviklet av GlaxoSmithKline og også utpekt et foreldreløst legemiddel av FDA, og ble godkjent av FDA 20. november 2008.
Trombopoietinreseptoragonister viste den største suksessen så langt i behandlingen av pasienter med ildfast ITP.
Bivirkninger av trombopoietinreseptoragonister inkluderer hodepine, ledd- eller muskelsmerter, svimmelhet, kvalme eller oppkast og økt risiko for blodpropp.
Kirurgi
Splenektomi (fjerning av milten ) kan vurderes hos pasienter som enten ikke reagerer på steroidbehandling, har hyppige tilbakefall eller ikke kan avta steroider etter noen måneder. Blodplater som er bundet av antistoffer blir tatt opp av makrofager i milten (som har Fc -reseptorer ), og dermed reduserer fjerning av milten ødeleggelse av blodplater. Prosedyren er potensielt risikabel i ITP -tilfeller på grunn av den økte muligheten for betydelig blødning under operasjonen. Varig remisjon etter splenektomi oppnås i 60 - 80 prosent av ITP -tilfellene. Selv om det er enighet om kortsiktig effekt av splenektomi, er funn om dets langsiktige effekt og bivirkninger kontroversielle. Etter splenektomi tilbakefall 11,6 - 75 prosent av ITP -tilfellene, og 8,7 - 40 prosent av ITP -tilfellene hadde ingen respons på splenektomi. Bruken av splenektomi for å behandle ITP har blitt mindre siden utviklingen av steroidterapi og andre farmasøytiske midler.
Trombocyttransfusjon
Trombocyttransfusjon alene anbefales normalt ikke bortsett fra i nødstilfeller, og det er vanligvis mislykket med å produsere en langsiktig blodplatetallstigning. Dette er fordi den underliggende autoimmune mekanismen som ødelegger pasientens blodplater også vil ødelegge donorplater, og derfor anses ikke blodplateoverføringer som et langsiktig behandlingsalternativ.
H. pylori -utryddelse
Hos voksne, spesielt de som bor i områder med høy forekomst av Helicobacter pylori (som normalt bor i mageveggen og har vært assosiert med magesår ), har identifisering og behandling av denne infeksjonen vist seg å forbedre antall blodplater hos en tredjedel av pasientene. I en femtedel normaliserte blodplatetallet seg fullstendig; denne svarprosenten er lik den som finnes ved behandling med rituximab, som er dyrere og mindre trygg. Hos barn støttes ikke denne tilnærmingen av bevis, bortsett fra i områder med høy forekomst. Urea -pustetesting og avføringsantigen -testing fungerer bedre enn serologi -baserte tester; dessuten kan serologi være falskt positiv etter behandling med IVIG.
Andre agenter
- Dapsone (også kalt difenylsulfon, DDS eller avlosulfon) er en anti-smittsom sulfonmedisin. Dapsone kan også være nyttig for behandling av lupus, revmatoid artritt og som en annenlinjebehandling for ITP. Mekanismen som dapson hjelper til med ITP er uklar, men et økt antall blodplater sees hos 40–60 prosent av mottakerne.
- Det off-label bruk av rituximab , et chimerisk monoklonalt antistoff mot den B-celle- overflateantigenet CD20 , kan det noen ganger være et effektivt alternativ til splenektomi. Imidlertid kan betydelige bivirkninger forekomme, og randomiserte kontrollerte studier er ikke avgjørende.
Prognose
Det er uvanlig at personer med ITP opplever alvorlig blødning (bare 5% av de som er berørt). Men innen fem år etter diagnosen er 15% av de berørte personene innlagt på sykehus med blødningskomplikasjoner.
Epidemiologi
Et normalt antall blodplater anses å være i området 150 000–450 000 per mikroliter (μl) blod for de fleste friske individer. Derfor kan man betraktes som trombocytopenisk under dette området, selv om terskelen for en diagnose av ITP ikke er knyttet til noe spesifikt tall.
Forekomsten av ITP er estimert til 50–100 nye tilfeller per million per år, med barn som står for halvparten av det antallet. Minst 70 prosent av barndomstilfellene vil ende med remisjon innen seks måneder, selv uten behandling. Videre vil en tredjedel av de gjenværende kroniske tilfellene vanligvis bli behandlet under oppfølgingsobservasjon, og en annen tredjedel vil ende opp med bare mild trombocytopeni (definert som blodplatetall over 50 000). En rekke immunrelaterte gener og polymorfismer har blitt identifisert som påvirker predisponering for ITP, med FCGR3a-V158-allel og KIRDS2/DL2 som øker mottakeligheten og KIR2DS5 er vist beskyttende.
ITP er vanligvis kronisk hos voksne, og sannsynligheten for varig remisjon er 20–40 prosent. Forholdet mellom menn og kvinner i voksengruppen varierer fra 1: 1,2 til 1,7 i de fleste aldersgrupper (barndomstilfeller er omtrent like store for begge kjønn), og medianalderen for voksne ved diagnosen er 56–60. Forholdet mellom mannlige og kvinnelige tilfeller hos voksne har en tendens til å øke med alderen. I USA antas den voksne kroniske befolkningen å være omtrent 60 000 - med kvinner som er flere enn 2 til 1, noe som har resultert i at ITP er blitt utpekt som en foreldreløs sykdom .
Den dødelighet på grunn av kronisk ITP varierer, men har en tendens til å være høyere i forhold til den generelle populasjonen for en hvilken som helst aldersgruppe. I en studie utført i Storbritannia ble det bemerket at ITP forårsaker en omtrent 60 prosent høyere dødelighet sammenlignet med kjønn- og alderssvarende personer uten ITP. Denne økte risikoen for død med ITP er i stor grad konsentrert til middelaldrende og eldre . Nittiseks prosent av de rapporterte ITP-relaterte dødsfallene var individer 45 år eller eldre. Det ble ikke observert noen signifikant forskjell i overlevelse blant menn og kvinner.
Svangerskap
Autoantistoffer mot blodplater hos en gravid kvinne med ITP vil angripe pasientens egne blodplater og vil også krysse morkaken og reagere mot fosterplater. Derfor er ITP en signifikant årsak til fostrets og nyfødte immuntrombocytopeni. Omtrent 10% av nyfødte som rammes av ITP vil ha blodplatetall <50.000/uL og 1% til 2% vil ha risiko for intracerebral blødning sammenlignbar med spedbarn med nyfødt alloimmun trombocytopeni (NAIT).
Ingen laboratorietester kan pålitelig forutsi om neonatal trombocytopeni vil forekomme. Risikoen for neonatal trombocytopeni er økt med:
- Mødre med en historie med splenektomi for ITP
- Mødre som hadde et tidligere spedbarn påvirket av ITP
- Graviditet (mors) blodplater teller mindre enn 100 000/uL
Det anbefales at gravide kvinner med trombocytopeni eller tidligere diagnose av ITP testes for serum trombocytantistoffer. En kvinne med symptomatisk trombocytopeni og et identifiserbart blodplate -antistoff bør startes på behandling for ITP, som kan omfatte steroider eller IVIG. Fosterblodanalyse for å bestemme antall blodplater utføres vanligvis ikke, da ITP-indusert trombocytopeni hos fosteret generelt er mindre alvorlig enn NAIT. Trombocyttransfusjon kan utføres hos nyfødte, avhengig av graden av trombocytopeni. Det anbefales at nyfødte blir fulgt med blodplatetall de første dagene etter fødselen.
Historie
Etter første rapporter fra den portugisiske legen Amato Lusitano i 1556 og Lazarus de la Rivière (lege til kongen av Frankrike) i 1658, var det den tyske legen og poeten Paul Gottlieb Werlhof som i 1735 skrev den mest fullstendige første rapporten om purpuraen til ITP. Blodplater var ukjente på den tiden. Navnet "Werlhofs sykdom" ble brukt mer mye før det nåværende beskrivende navnet ble mer populært. Blodplater ble beskrevet på begynnelsen av 1800 -tallet, og på 1880 -tallet koblet flere etterforskere purpuraen med abnormiteter i blodplatetallet. Den første rapporten om en vellykket terapi for ITP var i 1916, da en ung polsk medisinstudent, Paul Kaznelson , beskrev en kvinnelig pasients svar på en miltomi . Splenektomi forble et førstehjelpsmiddel frem til introduksjonen av steroidterapi på 1950-tallet.
Referanser
Eksterne linker
| Klassifisering | |
|---|---|
| Eksterne ressurser |