
Radikal polymerisering - Radical polymerization
Friradikalpolymerisasjon ( FRP ) er en polymerisasjonsmetode, ved hvilken en polymer dannes ved suksessiv tilsetning av frie radikaler . Frie radikaler kan dannes av en rekke forskjellige mekanismer, som vanligvis involverer separate initiatormolekyler. Etter generering tilfører den initierende frie radikalen (ikke-radikale) monomerenheter , og derved vokser polymerkjeden.
Friradikalpolymeriseringen er en nøkkel synteserute for å oppnå en rekke forskjellige polymerer og materialer kompositter . Den relativt uspesifikke naturen til kjemiske interaksjoner med frie radikaler gjør dette til en av de mest allsidige former for polymerisering tilgjengelig og muliggjør lette reaksjoner av polymere frie radikalkjedeender og andre kjemikalier eller underlag. I 2001 ble 40 milliarder av de 110 milliarder pund polymerer produsert i USA produsert ved fri-radikal polymerisering.
Friradikalpolymerisering er en type kjede-vekstpolymerisasjon , sammen med anionisk , kationisk og koordineringspolymerisasjon .
Innvielse
Initiering er det første trinnet i polymeriseringsprosessen . Under initiering opprettes et aktivt senter hvorfra en polymerkjede genereres. Ikke alle monomerer er utsatt for alle typer initiativtakere. Radikal initiering fungerer best på karbon-karbon dobbeltbindingen av vinylmonomere og karbon-oksygen dobbeltbinding i aldehyder og ketoner . Innvielsen har to trinn. I det første trinnet opprettes en eller to radikaler fra de initierende molekylene. I det andre trinnet overføres radikaler fra initiatormolekylene til de tilstedeværende monomerenhetene. Flere valg er tilgjengelige for disse initiativtakerne.
Typer av innvielse og initiativtakere
- Termisk nedbrytning
- Initiatoren oppvarmes til en binding spaltes homolytisk , og produserer to radikaler (figur 1). Denne metoden brukes oftest med organiske peroksider eller azoforbindelser .
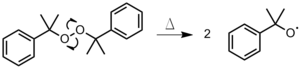 Figur 1 : Termisk dekomponering av di cumyl peroksyd
Figur 1 : Termisk dekomponering av di cumyl peroksyd - Fotolyse
- Stråling spalter en binding homolytisk, og produserer to radikaler (figur 2). Denne metoden brukes oftest med metalljodider, metallalkyler og azoforbindelser.Fotoinitiering kan også forekomme ved bi-molekylær H-abstraksjon når radikalen er i sin laveste triplet-eksiterte tilstand. Et akseptabelt fotoinitiatorsystem bør oppfylle følgende krav:

- Høy absorpsjonsevne i området 300–400 nm.
- Effektiv generering av radikaler som er i stand til å angripe den alken -dobbeltbindingen av vinylmonomere.
- Tilstrekkelig løselighet i bindemiddelsystemet ( prepolymer + monomer).
- Bør ikke gi guling eller ubehagelig lukt til det herdede materialet.
- Fotoinitiatoren og eventuelle biprodukter som oppstår ved bruk av den, skal være giftfri.
- redox reaksjoner
- Reduksjon av hydrogenperoksid eller et alkylhydrogenperoksid med jern (figur 3). Andre reduktanter slik som Cr2 + , V2 + , Ti3 + , Co2 + og Cu + kan i mange tilfeller anvendes i stedet for jernholdige ioner.
|
Figur 3 : Redoksreaksjon av hydrogenperoksid og jern.
|

- Persulfater
- Dissosiasjonen av et persulfat i den vandige fasen (figur 4). Denne metoden er nyttig i emulsjonspolymerisasjoner , der radikalen diffunderer til en hydrofob monomerholdig dråpe.
- Ioniserende stråling
- α- , β- , γ- eller røntgenstråler forårsaker utstøting av et elektron fra den initierende arten, etterfulgt av dissosiasjon og elektronfanging for å produsere en radikal (figur 5).
- Elektrokjemisk
- Elektrolyse av en løsning som inneholder både monomer og elektrolytt . Et monomermolekyl vil motta et elektron ved katoden for å bli et radikalanion, og et monomermolekyl vil gi opp et elektron ved anoden for å danne en radikal kation (figur 6). Radikalionene initierer deretter fri radikal (og / eller ionisk) polymerisering. Denne typen initiering er spesielt nyttig for å belegge metalloverflater med polymerfilmer.
- Plasma
- En gassformig monomer plasseres i elektrisk utladning ved lavt trykk under forhold der det dannes et plasma (ioniserte gassformige molekyler). I noen tilfeller blir systemet oppvarmet og / eller plassert i et radiofrekvensfelt for å hjelpe til med å skape plasma.
- Sonikering
- Høyintensiv ultralyd ved frekvenser utenfor området for menneskelig hørsel (16 kHz) kan påføres en monomer. Initiering skyldes effekter av kavitasjon (dannelse og kollaps av hulrom i væsken). Kollapsen av hulrommene genererer veldig høye lokale temperaturer og trykk. Dette resulterer i dannelsen av eksiterte elektroniske tilstander, som igjen fører til bindingsbrudd og radikal dannelse.
- Ternære initiativtakere
- En ternær initiativtaker er kombinasjonen av flere typer initiativtakere til ett initieringssystem. Typene initiatorer velges basert på egenskapene de er kjent for å indusere i polymerene de produserer. For eksempel har poly (metylmetakrylat) blitt syntetisert av det ternære systemet benzoylperoksid-3,6-bis ( o- karboksybenzoyl) - N- isopropylkarbazol-di-η 5 -indenylzikroniumdiklorid (figur 7).Denne typen initieringssystem inneholder et metallocen , en initiator og en heteroaromatisk diketo- karboksylsyre . Metallocener i kombinasjon med initiatorer akselererer polymerisering av poly (metylmetakrylat) og produserer en polymer med en smalere molekylvektfordeling. Eksemplet vist her består av indenylzirkonium (et metallocen) og benzoylperoksid (en initiator). Også initieringssystemer som inneholder heteroaromatiske diketo-karboksylsyrer, slik som 3,6-bis ( o- karboksybenzoyl) -N- isopropylkarbazol i dette eksempel, er kjent for å katalysere nedbrytningen av benzoylperoksyd. Initieringssystemer med denne spesielle heteroaromatiske diketkarboksylsyren er også kjent for å ha effekter på mikrostrukturen til polymeren. Kombinasjonen av alle disse komponentene - et metallocen, en initiator og en heteroaromatisk diketo-karboksylsyre - gir et ternært initieringssystem som ble vist å akselerere polymeriseringen og produsere polymerer med forbedret varmebestandighet og vanlig mikrostruktur.




Initiativtaker effektivitet
På grunn av bivirkninger og ineffektiv syntese av den radikale arten, er ikke kjedeinitiering 100% . Effektivitetsfaktoren f brukes til å beskrive den effektive radikale konsentrasjonen. Den maksimale verdien av f er 1, men typiske verdier varierer fra 0,3 til 0,8. Følgende er en liste over reaksjoner som reduserer effektiviteten til initiativtaker.
- Primær rekombinasjon
- To radikaler rekombineres igjen før en kjede setter i gang (figur 8). Dette skjer i løsningsmiddelburet , noe som betyr at det ikke har kommet noe løsningsmiddel mellom de nye radikalene.

- Andre rekombinasjonsveier
- To radikale initiatorer rekombinerer før de setter i gang en kjede, men ikke i løsemiddelburet (figur 9).
- Side reaksjoner
- Én radikal produseres i stedet for de tre radikale som kan produseres (figur 10).
|
Figur 10 : Reaksjon av polymerkjede R med andre arter i reaksjon
|

Formering
Under polymerisering bruker en polymer mesteparten av tiden på å øke kjedelengden, eller forplantes. Etter at den radikale initiativtaker er dannet, angriper den en monomer (figur 11). I en etenmonomer holdes et elektronpar sikkert mellom de to karbonene i en sigma-binding . Den andre holdes løst i en pi-obligasjon . Den frie radikalen bruker ett elektron fra pi-bindingen for å danne en mer stabil binding med karbonatomet. Det andre elektronet går tilbake til det andre karbonatomet, og gjør hele molekylet til en annen radikal. Dette begynner polymerkjeden. Figur 12 viser hvordan orbitalene til en etylenmonomer samhandler med en radikal initiator.


Når en kjede er startet, forplanter kjeden seg (figur 13) til det ikke er flere monomerer ( levende polymerisering ) eller til terminering skjer. Det kan være alt fra noen få til tusenvis av forplantningstrinn avhengig av flere faktorer som radikal reaksjon, kjedereaktivitet, løsningsmiddel og temperatur. Mekanismen for kjedeutbredelse er som følger:

Avslutning
Kjettingsterminering er uunngåelig ved radikal polymerisering på grunn av den høye reaktiviteten til radikaler. Oppsigelse kan skje med flere forskjellige mekanismer. Hvis lengre kjeder er ønsket, bør initiatorkonsentrasjonen holdes lav; Ellers vil mange kortere kjeder resultere.
- Kombinasjon av to aktive kjedeender: en eller begge av følgende prosesser kan forekomme.
- Kombinasjon: to kjedeender kobles ganske enkelt sammen for å danne en lang kjede (figur 14). Man kan bestemme om denne modusen for avslutning skjer ved å overvåke molekylvekten til den forplantende arten: kombinasjon vil resultere i en fordobling av molekylvekten. Dessuten vil kombinasjonen resultere i en polymer som er C to symmetrisk om punktet av kombinasjonen.
- Radikal disproporsjonering : et hydrogenatom fra en kjedeende blir abstrahert til en annen, og produserer en polymer med en terminal umettet gruppe og en polymer med en terminal mettet gruppe (figur 15).
- Kombinasjon av en aktiv kjedeende med en initiatorradikal (figur 16).
- Interaksjon med urenheter eller hemmere . Oksygen er den vanlige hemmeren. Den voksende kjeden vil reagere med molekylært oksygen, og produsere en oksygenradikal, som er mye mindre reaktiv (figur 17). Dette reduserer forplantningshastigheten betydelig.Nitrobensen , butylert hydroksyltoluen og difenylpikrylhydrazyl ( DPPH , figur 18) er noen få andre hemmere. Sistnevnte er en spesielt effektiv hemmer på grunn av resonansstabiliseringen av radikalet.





Kjedeoverføring
I motsetning til de andre modusene for avslutning, fører kjedeoverføring til ødeleggelse av bare en radikal, men også opprettelsen av en annen radikal. Ofte er imidlertid denne nyopprettede radikalen ikke i stand til videre forplantning. I likhet med disproporsjonering involverer alle kjedeoverføringsmekanismer også abstraksjon av et hydrogen eller annet atom. Det er flere typer kjedeoverføringsmekanismer.
- Til løsningsmiddel: et hydrogenatom blir abstrahert fra et løsningsmiddelmolekyl, noe som resulterer i dannelse av radikaler på løsemiddelmolekylene, som ikke vil forplante seg ytterligere (figur 19).Effektiviteten av kjedeoverføring som involverer løsemiddelmolekyler avhenger av mengden løsemiddel som er tilstede (mer løsemiddel fører til større sannsynlighet for overføring), styrken av bindingen som er involvert i abstraksjonstrinnet (svakere binding fører til større sannsynlighet for overføring), og stabiliteten av løsningsmiddelradikalet som dannes (større stabilitet fører til større sannsynlighet for overføring). Halogener , unntatt fluor , overføres lett.
- Til monomer: et hydrogenatom er abstrahert fra en monomer. Selv om dette skaper en radikal på den berørte monomeren, motvirker resonansstabilisering av denne radikalen ytterligere forplantning (figur 20).
-
Til initiator: en polymerkjede reagerer med en initiator, som avslutter den polymerkjeden, men skaper en ny radikal initiator (Figur 21). Denne initiatoren kan deretter starte nye polymerkjeder. Derfor, i motsetning til andre former for kjedeoverføring, tillater kjedeoverføring til initiativtakeren videre forplantning. Peroksidinitiatorer er spesielt følsomme for kjedeoverføring.

- Til polymer: radikalen i en polymerkjede trekker ut et hydrogenatom fra et sted i en annen polymerkjede (figur 22). Dette avslutter veksten av en polymerkjede, men lar den andre forgrene seg og gjenoppta veksten. Dette reaksjonstrinnet endrer verken antall polymerkjeder eller antall monomerer som har blitt polymerisert, slik at antallet gjennomsnittlig polymerisasjonsgrad er upåvirket.
Effekter av kjedeoverføring: Den mest åpenbare effekten av kjedeoverføring er en reduksjon i polymerkjedelengden. Hvis overføringshastigheten er mye større enn forplantningshastigheten, dannes veldig små polymerer med kjedelengder på 2-5 repeterende enheter ( telomerisering ). Mayo ligning anslår påvirkning av kjedeoverførings på kjedelengden ( x n ) . Der k tr er hastighetskonstanten for kjedeoverføring og k p er hastighetskonstanten for forplantning. Mayo-ligningen antar at overføring til løsningsmiddel er den viktigste avslutningsveien.
![{\ frac {1} {x_ {n}}} = \ venstre ({\ frac {1} {x_ {n}}} \ høyre) _ {o} + {\ frac {k _ {{tr}} [løsemiddel ]} {k_ {p} [monomer]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/3af10ecbdc45b856a8cd76ecc9502f63fc6bdc03)
Metoder
Det er fire industrielle metoder for radikal polymerisering:
- Bulkpolymerisasjon : reaksjonsblandingen inneholder bare initiator og monomer, uten løsemiddel.
- Løsningspolymerisasjon : reaksjonsblandingen inneholder løsningsmiddel, initiator og monomer.
- Suspensjonspolymerisasjon : reaksjonsblandingen inneholder en vandig fase, vannuoppløselig monomer og initiator løselig i monomerdråpene (både monomeren og initiatoren er hydrofob).
- Emulsjonspolymerisasjon : ligner suspensjonspolymerisasjon bortsett fra at initiatoren er løselig i den vandige fasen i stedet for i monomerdråpene (monomeren er hydrofob, og initiatoren er hydrofil). Et emulgeringsmiddel er også nødvendig.
Andre metoder for radikal polymerisering inkluderer følgende:
- Malpolymerisering : I denne prosessen får polymerkjeder vokse langs malmakromolekyler for det meste av livet. En velvalgt mal kan påvirke polymerisasjonshastigheten så vel som molær masse og mikrostruktur til datterpolymeren. Molmassen til en datterpolymer kan være opptil 70 ganger større enn den for polymerer produsert i fravær av mal og kan ha høyere molmasse enn malene selv. Dette er på grunn av retardasjon av avslutningen for malassosierte radikaler og ved å hoppe av en radikal til nabomalen etter å ha nådd slutten av en malpolymer.
- Plasmapolymerisering : Polymeriseringen initieres med plasma. En rekke organiske molekyler inkludert alkener , alkyner og alkaner gjennomgår polymerisering til produkter med høy molekylvekt under disse betingelser. Forplantningsmekanismene ser ut til å involvere både ioniske og radikale arter. Plasmapolymerisering tilbyr en potensielt unik metode for å danne tynne polymerfilmer for bruk som tynne filmkondensatorer, antirefleksbelegg og forskjellige typer tynne membraner.
- Sonikering : Polymeriseringen initieres av ultralyd med høy intensitet. Polymerisering til polymer med høy molekylvekt observeres, men konverteringene er lave (<15%). Polymerisasjonen er selvbegrensende på grunn av den høye viskositeten som produseres selv ved lav konvertering. Høy viskositet hindrer kavitasjon og radikal produksjon.
Reversibel deaktiveringsradikal polymerisering
Også kjent som levende radikalpolymerisasjon , kontrollert radikalpolymerisasjon, reversibel deaktiveringsradikalpolymerisasjon (RDRP) er avhengig av helt rene reaksjoner, og forhindrer avslutning forårsaket av urenheter. Fordi disse polymerisasjonene bare stopper når det ikke er mer monomer, kan polymerisasjonen fortsette etter tilsetning av mer monomer. Blokk kopolymerer kan lages på denne måten. RDRP muliggjør kontroll av molekylvekt og spredning. Dette er imidlertid veldig vanskelig å oppnå, og i stedet opptrer en pseudolevende polymerisering med bare delvis kontroll av molekylvekt og dispersitet. ATRP og RAFT er hovedtyper av fullstendig radikal polymerisering.
- Atom overføring radikal polymerisering (ATRP): basert på dannelse av en karbon-karbonbinding for atom overføring radikal tilsetning. Denne metoden, uavhengig oppdaget i 1995 av Mitsuo Sawamoto og av Jin-Shan Wang og Krzysztof Matyjaszewski , krever reversibel aktivering av en sovende art (for eksempel et alkylhalogenid ) og en overgangsmetallhalogenidkatalysator (for å aktivere sovende arter).
- Reversibel addisjon-fragmentering kjedeoverføringspolymerisering (RAFT): krever en forbindelse som kan fungere som et reversibelt kjedeoverføringsmiddel, slik som ditioforbindelse.
- Stabil fri radikal polymerisering (SFRP) : brukes til å syntetisere lineære eller forgrenede polymerer med smale molekylvektfordelinger og reaktive endegrupper på hver polymerkjede. Prosessen har også blitt brukt til å lage blokk-sampolymerer med unike egenskaper. Konverteringsfrekvensen er omtrent 100% ved bruk av denne prosessen, men krever temperaturer på ca. 135 ° C. Denne prosessen er mest brukt med akrylater, styrener og diener. Reaksjonsskjemaet i figur 23 illustrerer SFRP-prosessen. Fordi kjedeenden er funksjonalisert med TEMPO- molekylet (Figur 24), reduseres for tidlig avslutning ved kobling. Som med alle levende polymerisasjoner, vokser polymerkjeden til hele monomeren er forbrukt.


Kinetikk
I typiske kjedevekstpolymerisasjoner kan reaksjonshastighetene for initiering, forplantning og avslutning beskrives som følger:
![v_ {i} = {\ operatorname {d} [M \ cdot] / \ operatorname {d} t} = 2k_ {d} f [I]](https://wikimedia.org/api/rest_v1/media/math/render/svg/f02573160d99ce28cd15dfe6c9f111e9c47c082d)
![v_ {p} = k_ {p} [M] [M \ cdot]](https://wikimedia.org/api/rest_v1/media/math/render/svg/bc7bb155b647c8e9a58d7d300e71a52898b8f221)
![v_ {t} = {- \ operatorname {d} [M \ cdot] / \ operatorname {d} t} = 2k_ {t} [M \ cdot] ^ {2}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e451f440b60e6b1e9fbe627b99a0d6eb734aeea4)
hvor f er effektiviteten til initiatoren og k d , k p og k t er konstanter for henholdsvis initiator dissosiasjon, kjedeutbredelse og avslutning. [I] [M] og [M •] er konsentrasjonene av initiatoren, monomeren og den aktivt voksende kjeden.
Under steady-state-tilnærmingen forblir konsentrasjonen av de aktivt voksende kjedene konstant, dvs. at hastighetene for initiering og avslutning er like. Konsentrasjonen av aktiv kjede kan avledes og uttrykkes som andre kjente arter i systemet.
![[M \ cdot] = \ left ({\ frac {k_ {d} [I] f} {k_ {t}}} \ right) ^ {{1/2}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/92af6e128be7fa6a566a3590c6f267e601e5e3e2)
I dette tilfellet kan hastigheten på kjedeutbredelse beskrives nærmere ved anvendelse av en funksjon av initiator- og monomerkonsentrasjonen
![{\ displaystyle v_ {p} = {k_ {p}} \ left ({\ frac {fk_ {d}} {k_ {t}}} \ right) ^ {1/2} [I] ^ {1/2 } [M]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8fab2227059300c21a6173d5108ca221645da4cf)
Den kinetiske kjedelengden v er et mål på gjennomsnittlig antall monomerenheter som reagerer med et aktivt senter i løpet av dets levetid og er relatert til molekylvekten gjennom termineringsmekanismen. Uten kjedeoverføring er den kinetiske kjedelengden bare en funksjon av forplantningshastighet og initieringshastighet.
![{\ displaystyle \ nu = {\ frac {v_ {p}} {v_ {i}}} = {\ frac {k_ {p} [M] [M \ cdot]} {2fk_ {d} [I]}} = {\ frac {k_ {p} [M]} {2 (fk_ {d} k_ {t} [I]) ^ {1/2}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/60318ebbc0960f885949ea451e3692f7cebae026)
Forutsatt ingen kjedeoverføringseffekt oppstår i reaksjonen, den antallsmidlere polymerisasjonsgrad P n kan korreleres med den kinetiske kjedelengde. Ved avslutning ved disproporsjonering produseres ett polymermolekyl per hver kinetiske kjede:

Terminering ved kombinasjon fører til ett polymermolekyl per to kinetiske kjeder:

Enhver blanding av begge disse mekanismene kan beskrives ved å bruke verdien δ , bidraget til disproporsjonering til den totale avslutningsprosessen:

Hvis kjedeoverføring vurderes, påvirkes ikke den kinetiske kjedelengden av overføringsprosessen fordi det voksende frie radikalsenteret generert av initieringstrinnet holder seg i live etter en hvilken som helst kjedeoverføringshendelse, selv om det produseres flere polymerkjeder. Imidlertid synker antall gjennomsnittlig polymerisasjonsgrad når kjedeoverføringene, siden de voksende kjedene avsluttes av kjedeoverføringshendelsene. Tatt i betraktning den kjedeoverføringsreaksjon mot løsemiddel S , initiator I , polymer P , og det tilsatt kjedeoverføringsmiddel T . Likningen for P n vil bli endret som følger:
![{\ displaystyle {\ frac {1} {x_ {n}}} = {\ frac {2k_ {t, d} + k_ {t, c}} {{k_ {p}} ^ {2} [M] ^ {2}}} v_ {p} + C_ {M} + C_ {S} {\ frac {[S]} {[M]}} + C_ {I} {\ frac {[I]} {[M] }} + C_ {P} {\ frac {[P]} {[M]}} + C_ {T} {\ frac {[T]} {[M]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f87872861546ef6fc87a203d04693d6898ca25d2)
Det er vanlig å definere kjedeoverføringskonstanter C for de forskjellige molekylene
- , , , ,





Termodynamikk
I kjedevekstpolymerisasjon kan posisjonen til likevekt mellom polymer og monomerer bestemmes av termodynamikken til polymerisasjonen. Den Gibbs fri energi (Ag p ) av polymeriseringen er vanligvis brukes til å kvantifisere tendensen hos en polymer reaksjon. Polymerisasjonen vil være favorisert hvis ΔG p <0; hvis ΔG p > 0, vil polymeren gjennomgå depolymerisering . I henhold til den termodynamiske ligningen ΔG = ΔH - TΔS, vil en negativ entalpi og en økende entropi forskyve likevekten mot polymerisering.
Generelt er polymeriseringen en eksoterm prosess, dvs. negativ entalpiendring , siden tilsetning av en monomer til den voksende polymerkjeden innebærer konvertering av π-bindinger til σ-bindinger, eller en ringåpningsreaksjon som frigjør ringspenningen i en syklisk monomer. . I mellomtiden, under polymerisering, er en stor mengde små molekyler assosiert, og taper rotasjon og translasjonelle frihetsgrader . Som et resultat avtar entropien i systemet, AS p <0 for nesten alle polymeriseringsprosesser. Siden depolymerisering nesten alltid er entropisk favorisert, må Ah p da være tilstrekkelig negativ til å kompensere for den ugunstige entropiske betegnelsen. Først da vil polymerisering favoriseres termodynamisk av den resulterende negative AG p .
I praksis utføres polymer favoriseres ved lave temperaturer: TΔS p er liten. Depolymerisering foretrekkes ved høye temperaturer: TΔS p er stor. Når temperaturen øker, blir ΔG p mindre negativ. Ved en viss temperatur når polymerisasjonen likevekt (polymerisasjonshastighet = depolymerisasjonshastighet). Denne temperaturen kalles taktemperaturen ( Tc ). ΔG p = 0.
Stereokjemi
Stereokjemien ved polymerisering er opptatt av forskjellen i atomforbindelse og romlig orientering i polymerer som har samme kjemiske sammensetning.
Hermann Staudinger studerte stereoisomerismen i kjedepolymerisasjon av vinylmonomerer på slutten av 1920-tallet, og det tok ytterligere to tiår før folk fullt ut satte pris på ideen om at hvert av formeringstrinnene i polymerveksten kunne gi opphav til stereoisomerisme. Den viktigste milepælen i stereokjemi ble etablert av Ziegler og Natta og deres kolleger på 1950-tallet, da de utviklet metallbasert katalysator for å syntetisere stereoregulære polymerer. Årsaken til at stereokjemien til polymeren er av spesiell interesse, er at den fysiske oppførselen til en polymer ikke bare avhenger av den generelle kjemiske sammensetningen, men også av de mer subtile forskjellene i mikrostruktur . Ataktiske polymerer består av et tilfeldig arrangement av stereokjemi og er amorfe (ikke-krystallinske), myke materialer med lavere fysisk styrke. De tilsvarende isotaktiske (som substituenter alle på samme side) og syndiotaktiske (som substituenter av alternative repeterende enheter på samme side) polymerer oppnås vanligvis som høykrystallinske materialer. Det er lettere for stereoregulære polymerer å pakke inn i et krystallgitter siden de er mer ordnet og den resulterende krystalliniteten fører til høyere fysisk styrke og økt løsemiddel- og kjemisk motstand samt forskjeller i andre egenskaper som er avhengig av krystallinitet. Det viktigste eksemplet på den industrielle bruken av stereoregulære polymerer er polypropen . Isotaktisk polypropen er en høysmeltende (165 ° C), sterk, krystallinsk polymer, som brukes både som plast og fiber. Ataktisk polypropen er et amorft materiale med et oljeaktig til voksaktig mykt utseende som finner bruk i asfaltblandinger og formuleringer for smøremidler, tetningsmidler og lim, men volumene er små sammenlignet med isotaktisk polypropen.
Når en monomer tilfører en radikal kjedeende, er det to faktorer å ta hensyn til når det gjelder stereokjemi: 1) samspillet mellom den terminale kjedekarbonet og det monomermolekylet som nærmer seg og 2) konfigurasjonen av den nest siste repeterende enheten i polymerkjeden. Det terminale karbonatomet har sp 2- hybridisering og er plan. Vurder polymeriseringen av monomeren CH 2 = CXY. Det er to måter et monomermolekyl kan nærme seg det terminale karbonet på: speiltilnærmingen (med like substituenter på samme side) eller ikke-speiltilnærmingen (som substituenter på motsatte sider). Hvis fri rotasjon ikke forekommer før neste monomer legger til, vil speiltilnærmingen alltid føre til en isotaktisk polymer, og ikke-speiltilnærmingen vil alltid føre til en syndiotaktisk polymer (figur 25).

Imidlertid, hvis interaksjoner mellom substituentene til den nest siste repeterende enheten og det terminale karbonatomet er signifikante, kan konformasjonsfaktorer føre til at monomeren tilfører polymeren på en måte som minimerer sterisk eller elektrostatisk interaksjon (figur 26).

Reaktivitet
Tradisjonelt blir reaktiviteten til monomerer og radikaler vurdert ved hjelp av kopolymeriseringsdata . Den Q-e ordningen, den mest brukte verktøy for semi-kvantitativ forutsigelse av monomer reaktivitetsforhold , ble først foreslått av Alfrey og pris i 1947. Den forbindelse skal det tas hensyn til den iboende termodynamiske stabilitet og polare effekter i overgangstilstanden. En gitt radikal og en monomer som er vurdert å ha iboende reaktivitet P I og Q j , henholdsvis. De polare effektene i overgangstilstanden, den antatte permanente elektriske ladningen som bæres av den enheten (radikal eller molekyl), blir kvantifisert av faktoren e , som er en konstant for en gitt monomer, og har samme verdi for radikalen avledet fra den spesifikk monomer. For tilsetningen av monomer 2 til en voksende polymerkjede hvis aktive ende er resten av monomeren 1, hastighetskonstanten, k 12 , er antatt å være relatert til de fire relevante parametere reaktivitet av



Monomerreaktivitetsforholdet for tilsetning av monomerer 1 og 2 til denne kjeden er gitt av

For kopolymerisering av et gitt par monomerer tillater de to eksperimentelle reaktivitetsforholdene r 1 og r 2 evaluering av (Q 1 / Q 2 ) og (e 1 - e 2 ). Verdier for hver monomer kan deretter tildeles i forhold til en referansemonomer, vanligvis valgt som styren med vilkårlige verdier Q = 1.0 og e = –0.8.
applikasjoner
Fri radikalpolymerisering har funnet anvendelser inkludert produksjon av polystyren , termoplastiske blokkopolymerelastomerer , kardiovaskulære stenter , kjemiske overflateaktive stoffer og smøremidler. Blokk-kopolymerer brukes til en lang rekke bruksområder, inkludert lim, fottøy og leker.
Fri radikalpolymerisering har også bruk i forskning, for eksempel i funksjonalisering av karbonnanorør . CNTs iboende elektroniske egenskaper får dem til å danne store aggregater i løsning, og utelukker nyttige applikasjoner. Å legge til små kjemiske grupper på veggene i CNT kan eliminere denne tilbøyeligheten og justere responsen til det omgivende miljøet. Bruk av polymerer i stedet for mindre molekyler kan endre CNT-egenskaper (og omvendt kan nanorør endre polymermekaniske og elektroniske egenskaper). For eksempel belagte forskere karbonnanorør med polystyren ved først å polymerisere polystyren via kjederadikalpolymerisasjon og deretter blande det ved 130 ° C med karbonnanorør for å generere radikaler og pode dem på veggene i karbonnanorør (figur 27). Kjedevekstpolymerisering ("poding til") syntetiserer en polymer med forhåndsbestemte egenskaper. Rensing av polymeren kan brukes til å oppnå en mer jevn lengdefordeling før poding. Omvendt, “pode på”, med radikal-polymerisasjonsmetoder som atom overføring radikal polymerisering (ATRP) eller nitroksid-mediert polymerisering (NMP), tillater hurtig vekst av høymolekylære polymerer.

Radikal polymerisering hjelper også til syntese av nanokompositt hydrogeler . Disse gelene er laget av vannsvellbar nanoskala leire (spesielt de som er klassifisert som smektitter ) innhyllet av en nettverkspolymer . De er ofte biokompatible og har mekaniske egenskaper (som fleksibilitet og styrke) som lover applikasjoner som syntetisk vev. Syntese innebærer polymerisering av frie radikaler. Den generelle synteseprosedyren er avbildet i figur 28. Leire er dispergert i vann, hvor den danner veldig små, porøse plater. Deretter tilsettes initiatoren og en katalysator, etterfulgt av tilsetning av den organiske monomeren, generelt et akrylamid- eller akrylamidderivat. Initiativtakeren er valgt for å ha sterkere interaksjon med leiren enn den organiske monomeren, slik at den fortrinnsvis adsorberes til leireoverflaten. Blandingen og vannløsningsmidlet oppvarmes for å starte polymerisasjon. Polymerer vokser av initiatorene som igjen er bundet til leire. På grunn av rekombinasjons- og disproporsjoneringsreaksjoner, binder voksende polymerkjeder til hverandre og danner en sterk, tverrbundet nettverkspolymer, med leirepartikler som fungerer som forgreningspunkter for flere polymerkjedesegmenter. Fri radikalpolymerisasjon brukt i denne sammenheng tillater syntese av polymerer fra et bredt utvalg av substrater (kjemikaliene til passende leire varierer). Termineringsreaksjoner unike for kjedevekstpolymerisering produserer et materiale med fleksibilitet, mekanisk styrke og biokompatibilitet.

Elektronikk
Det radikale polymerglasset PTMA er omtrent 10 ganger mer elektrisk ledende enn vanlige halvledende polymerer. PTMA er i en klasse med elektrisk aktive polymerer som kan finne bruk i gjennomsiktige solceller , antistatiske og antiglansbelegg for mobiltelefonskjermer , antistatiske belegg for fly for å beskytte mot lynnedslag, fleksible flash-stasjoner og termoelektriske enheter, som konverterer strøm til varme og omvendt. Utbredte praktiske bruksområder krever økt ledningsevne ytterligere 100 til 1000 ganger.
Polymeren ble opprettet ved hjelp av avbeskyttelse, som innebærer å erstatte et spesifikt hydrogenatom i anhengsgruppen med et oksygenatom. Det resulterende oksygenatomet i PTMA har ett uparret elektron i det ytre skallet, noe som gjør det mottakelig for transportladning. Avbeskyttelsestrinnet kan føre til fire forskjellige kjemiske funksjoner, hvorav to er lovende for å øke ledningsevnen.
Se også
- Anionisk tilleggspolymerisering
- Kjede-vekst polymerisering
- Kjedeoverføring
- Koboltmediert radikal polymerisering
- Levende polymerisering
- Nitroksid-mediert radikal polymerisering
- Polymer
- Polymerisering
- Reversibel-deaktivering radikal polymerisering
- Trinnvekstpolymerisering
Referanser
Eksterne linker
- Tilleggspolymerisering
- Gratis radikal polymerisering (videoanimasjon)
- Gratis radikal polymerisering - kjedeoverføring
- Gratis radikal vinylpolymerisering
- Polymeriseringen av Alkenes
- Polymer syntese
- Radical Reaction Chemistry
- Stabil fri radikal polymerisering