
Wilms svulst - Wilms' tumor
| Wilms svulst | |
|---|---|
| Andre navn | Wilms svulst Nephroblastoma |
 | |
| Mikrofotografi med høy forstørrelse som viser de tre elementene i Wilms svulst. H & E flekk . | |
| Uttale | |
| Spesialitet | Onkologi , urologi , nefrologi |
| Behandling | Nefrektomi Strålebehandling |
Wilms svulst , også kjent som nefroblastom , er en kreft i nyrene som vanligvis forekommer hos barn , sjelden hos voksne . Den er oppkalt etter Max Wilms , den tyske kirurgen (1867–1918) som først beskrev den.
Omtrent 650 tilfeller blir diagnostisert i USA årlig. Flertallet av tilfellene forekommer hos barn uten tilknyttede genetiske syndromer; Imidlertid har et mindretall av barn med Wilms svulst en medfødt abnormitet. Det reagerer veldig på behandling, med om lag 9/10 barn som blir kurert.
Tegn og symptomer
Typiske tegn og symptomer på Wilms svulst inkluderer følgende:
- en smertefri, håndgripelig magemasse
- tap av Appetit
- magesmerter
- feber
- kvalme og oppkast
- blod i urinen (i omtrent 20% av tilfellene)
- høyt blodtrykk i noen tilfeller (spesielt hvis synkron eller metakron bilateral nyreinnblanding)
- Sjelden som varicocele
Patogenese


Wilms svulst har mange årsaker, som stort sett kan kategoriseres som syndromisk og ikke-syndromisk. Syndromiske årsaker til Wilms svulst oppstår som et resultat av endringer i gener som Wilms Tumor 1 (WT1) eller Wilms Tumor 2 (WT2) gener, og svulsten presenterer en gruppe andre tegn og symptomer. Ikke-syndromisk Wilms svulst er ikke forbundet med andre symptomer eller patologier. Mange, men ikke alle tilfeller av Wilms svulst utvikler seg fra nefrogen hviler, som er fragmenter av vev i eller rundt nyrene som utvikler seg før fødselen og blir kreftsykre etter fødselen. Spesielt er tilfeller av bilateral Wilms svulst, så vel som tilfeller av Wilms svulst avledet fra visse genetiske syndromer som Denys-Drash syndrom , sterkt forbundet med nefrogen hvile. De fleste nefroblastomer er bare på den ene siden av kroppen og finnes på begge sider i mindre enn 5% av tilfellene, selv om personer med Denys-Drash syndrom for det meste har bilaterale eller flere svulster. De pleier å være innkapslede og vaskulariserte svulster som ikke krysser midtlinjen i magen. I tilfeller av metastase er det vanligvis til lungen. Et brudd i Wilms svulst setter pasienten i fare for blødning og peritoneal spredning av svulsten. I slike tilfeller er kirurgisk inngrep av en kirurg som har erfaring med fjerning av en så skjør svulst avgjørende.
Patologisk består et trifasisk nefroblastom av tre elementer:
Wilms svulst er en ondartet svulst som inneholder metanephric blastema , stromale og epitelivater. Karakteristisk er tilstedeværelsen av abortive tubuli og glomeruli omgitt av et spindlet cellestroma. Stroma kan omfatte striert muskel , brusk , bein , fettvev og fibrøst vev. Dysfunksjon er forårsaket når svulsten komprimerer det normale nyreparenkymet.
Den mesenkymale komponenten kan omfatte celler som viser rabdomyoid differensiering eller malignitet ( rabdomyosarkomatiske Wilms).
Wilms svulster kan skilles i to prognostiske grupper basert på patologiske egenskaper:
- Gunstig - Inneholder velutviklede komponenter som er nevnt ovenfor
- Anaplastisk - Inneholder diffus anaplasi (dårlig utviklede celler)
Mutasjoner av WT1 -genet som er lokalisert på den korte armen til kromosom 11 (11p13) observeres i omtrent 20% av Wilms svulster, hvorav de fleste arves fra kimlinjen , mens et mindretall ervervet somatiske mutasjoner . I tillegg bærer minst halvparten av Wilms svulster med mutasjoner i WT1 også ervervede somatiske mutasjoner i CTNNB1 , genet som koder for proto-onkogenet beta-catenin . Dette sistnevnte genet finnes på den korte armen til kromosom 3 (3p22.1).
De fleste tilfeller har ikke mutasjoner i noen av disse genene.
| Syndromnavn | Tilknyttet genetisk variant | Risiko for Wilms svulst | Beskrivelse av syndrom |
| WAGR syndrom (Wilms tumor, aniridia, kjønnsanomalier, retardasjon) | Gensletting som inkluderer både WT1 og PAX6 | 45–60% | Karakterisert ved Wilms tumor, aniridia (fravær av iris), hemihypertrophy (ene side av legemet er større enn den andre), urin abnormiteter, tvetydige genitalia, utviklingshemming. |
| Denys-Drash syndrom (DDS) | WT1 (ekson 8 og 9) | 74% | Karakterisert av nyresykdommer siden fødselen førte til tidlig nyresvikt, tvetydige kjønnsorganer (interseksuelle lidelser). |
| Beckwith-Wiedemann syndrom | Unormal regulering av kromosom 11p15.5 | 7% | Karakterisert av makrosmia (stor fødselsstørrelse), makroglossi (stor tunge), hemihypertrofi (den ene siden av kroppen er større), andre svulster i kroppen, omphalocele (åpen bukvegg) og visceromegali (forstørrelse av organer inne i magen). |
Det er rapportert en tilknytning til H19 . H19 er et langt ikke -kodende RNA plassert på den korte armen til kromosom 11 (11p15.5).
Diagnose
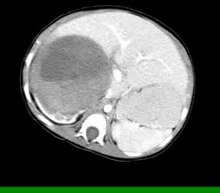
Flertallet av mennesker med Wilms svulst har en asymptomatisk magemasse som blir lagt merke til av et familiemedlem eller helsepersonell. Nyresvulster kan også bli funnet under rutinemessig screening hos barn som har kjent predisponerende kliniske syndromer. Diagnostikkprosessen inkluderer å ta en medisinsk historie, en fysisk eksamen og en rekke tester, inkludert blod-, urin- og bildebehandlingstester.
Når Wilms svulst mistenkes, gjøres vanligvis en ultralydsskanning først for å bekrefte tilstedeværelsen av en intrarenal masse. En datortomografisk skanning eller MR -skanning kan også brukes for mer detaljert bildebehandling. Til slutt bekreftes diagnosen Wilms svulst av en vevsprøve. I de fleste tilfeller utføres ikke en biopsi først fordi det er fare for at kreftceller sprer seg under prosedyren. Behandling i Nord -Amerika er nefrektomi eller i Europa kjemoterapi etterfulgt av nefrektomi. En definitiv diagnose oppnås ved patologisk undersøkelse av nefrektomi -prøven.
Iscenesettelse
Staging er en standard måte å beskrive omfanget av spredning av Wilms svulster og å bestemme prognose og behandlinger. Staging er basert på anatomiske funn og patologi av tumorceller. I henhold til omfanget av tumorvev på tidspunktet for første diagnose, vurderes fem stadier.
I svulst i Ims Wilms (43% av tilfellene) må alle følgende kriterier være oppfylt:
- Svulst er begrenset til nyrene og er fullstendig fjernet.
- Overflaten på nyrekapslen er intakt.
- Svulsten er ikke sprukket eller biopsiert (åpen eller nål) før fjerning.
- Ingen involvering av ekstrarenal eller renal sinus lymfekar
- Ingen gjenværende svulst er synlig utover eksisjonens marginer.
- Metastase av svulst til lymfeknuter ikke identifisert.
I trinn II (23% av tilfellene) må 1 eller flere av følgende kriterier være oppfylt:
- Svulst strekker seg utover nyrene, men er fullstendig utskåret.
- Ingen gjenværende svulst er synlig ved eller utenfor grensene for eksisjon.
- En av følgende betingelser kan også eksistere:
- Tumorinnblanding av blodårene i nyresinus og/eller utenfor nyreparenchymet.
- Omfattende tumorinnblanding av renal sinus bløtvev.
I trinn III (20% av tilfellene) må 1 eller flere av følgende kriterier være oppfylt:
- Inoperabel primær svulst.
- Metastase i lymfeknuter.
- Svulst er tilstede ved kirurgiske marginer.
- Svulstsøl som involverer peritoneale overflater, enten før eller under operasjonen, eller transeksjonert tumortromb.
- Svulsten har blitt biopsiert før fjerning eller det er lokalt søl av svulst under operasjonen, begrenset til flanken.
Fase IV (10% av tilfellene) Wilms svulst er definert av tilstedeværelsen av hematogene metastaser (lunge, lever, bein eller hjerne), eller lymfeknute -metastaser utenfor bukoperasjonen.
Fase V (5% av tilfellene) Wilms svulst er definert av bilateral nyreinnblanding på tidspunktet for første diagnose. For pasienter med bilateral involvering, bør det forsøkes å trinnvise hver side i henhold til kriteriene ovenfor (trinn I til III) på grunnlag av sykdomsomfang før biopsi.
Behandling og prognose
Den totale 5-års overlevelsen er estimert til å være omtrent 90%, men for individer er prognosen sterkt avhengig av individuell iscenesettelse og behandling . Tidlig fjerning har en tendens til å fremme positive resultater.
Tumorspesifikk tap av heterozygositet (LOH) for kromosomer 1p og 16q identifiserer en delmengde av Wilms svulstpasienter som har en betydelig økt risiko for tilbakefall og død. LOH for disse kromosomale regionene kan nå brukes som en uavhengig prognostisk faktor sammen med sykdomsstadiet for å målrette intensiteten av behandlingen mot risiko for behandlingssvikt. Genomeomfattende kopienummer og LOH-status kan vurderes med virtuell karyotyping av tumorceller (ferske eller parafininnstøpte).
Noen ganger kan statistikk vise gunstigere utfall for mer aggressive stadier enn for mindre aggressive stadier, som kan skyldes mer aggressiv behandling og/eller tilfeldig variasjon i studiegruppene. Et stadium V -svulst er ikke nødvendigvis verre enn et stadium IV -svulst.
| Scene | Histopatologi | 4 års tilbakefallsfri overlevelse (RFS) eller hendelsesfri overlevelse (EFS) | 4 års total overlevelse (OS) | Behandling |
|---|---|---|---|---|
| Fase I | Gunstig histologi hos barn yngre enn 24 måneder eller tumorvekt mindre enn 550g | 85% | 98% | Bare kirurgi (bør bare utføres i forbindelse med en klinisk prøve) |
| Gunstig histologi hos barn eldre enn 24 måneder eller tumorvekt over 550g | 94% RFS | 98% | Nefrektomi + prøvetaking av lymfeknuter etterfulgt av regime EE-4A | |
| Diffus anaplastisk | 68% EFS | 80% | Nefrektomi + prøvetaking av lymfeknuter etterfulgt av regime EE-4A og strålebehandling | |
| Fase II | Gunstig histologi | 86% RFS | 98% | Nefrektomi + prøvetaking av lymfeknuter etterfulgt av regime EE-4A |
| Fokal anaplastisk | 80% EFS | 80% | Nefrektomi + prøvetaking av lymfeknuter etterfulgt av strålebehandling i magen og regime DD-4A | |
| Diffus anaplastisk | 83% EFS | 82% | Nefrektomi + prøvetaking av lymfeknuter etterfulgt av abdominal strålebehandling og diett I | |
| Trinn III | Gunstig histologi | 87% RFS | 94% | Nefrektomi + prøvetaking av lymfeknuter etterfulgt av strålebehandling i magen og regime DD-4A |
| Fokal anaplastisk | 88% RFS | 100% (8 personer i studiet) | Nefrektomi + prøvetaking av lymfeknuter etterfulgt av strålebehandling i magen og regime DD-4A | |
| Fokal anaplastisk (preoperativ behandling) | 71% RFS | 71% | Preoperativ behandling med regime DD-4A etterfulgt av nefrektomi + prøvetaking av lymfeknuter og strålebehandling i magen | |
| Diffus anaplastisk | 46% EFS | 53% | Preoperativ behandling med diett I etterfulgt av nefrektomi + prøvetaking av lymfeknuter og strålebehandling i magen | |
| Diffus anaplastisk | 65% EFS | 67% | Umiddelbar nefrektomi + prøvetaking av lymfeknuter etterfulgt av strålebehandling i magen og diett I | |
| Fase IV | Gunstig histologi | 76% RFS | 86% | Nefrektomi + prøvetaking av lymfeknuter, etterfulgt av abdominal strålebehandling, bilateral lungestrålebehandling og regime DD-4A |
| Fokal anaplastisk | 61% EFS | 72% | Nefrektomi + prøvetaking av lymfeknuter, etterfulgt av abdominal strålebehandling, bilateral lungestrålebehandling og regime DD-4A | |
| Diffus anaplastisk | 33% EFS | 33% | Umiddelbar nefrektomi + prøvetaking av lymfeknuter etterfulgt av strålebehandling i magen, strålebehandling med full lunge og diett I | |
| Diffus anaplastisk (preoperativ behandling) | 31% EFS | 44% | Preoperativ behandling med diett I etterfulgt av nefrektomi + prøvetaking av lymfeknuter etterfulgt av strålebehandling i magen, strålebehandling med full lunge | |
| Fase V | Alt i alt | 61% EFS | 80% | |
| Gunstig histologi | 65% | 87% | Preoperativ behandling med regime DD-4A , etterfulgt av nefronbesparende kirurgi eller nefrekomi, iscenesettelse av svulster og cellegift og/eller strålebehandling basert på patologi og iscenesettelse | |
| Fokal anaplastisk | 76% | 88% | Preoperativ behandling med regime DD-4A , etterfulgt av nefronbesparende kirurgi eller nefrekomi, iscenesettelse av svulster og cellegift og/eller strålebehandling basert på patologi og iscenesettelse | |
| Diffus anaplastisk | 25% | 42% | Preoperativ behandling med regime DD-4A , etterfulgt av nefronbesparende kirurgi eller nefrekomi, iscenesettelse av svulster og cellegift og/eller strålebehandling basert på patologi og iscenesettelse |
Ved tilbakefall av Wilms svulst, har 4-års overlevelsesrate for barn med standardrisiko blitt estimert til å være 80%.
Epidemiologi
Wilms -svulst er den vanligste maligne nyretumoren hos barn. Det er en rekke sjeldne genetiske syndromer som har blitt knyttet til økt risiko for å utvikle Wilms Tumor. Screening -retningslinjene varierer fra land til land; helsepersonell anbefaler imidlertid regelmessig ultralydundersøkelse for personer med tilhørende genetiske syndromer.
Wilms svulst påvirker omtrent én person per 10 000 på verdensbasis før fylte 15 år. Personer med afrikansk avstamning kan ha litt høyere forekomst av Wilms svulst. Toppalderen for Wilms svulst er 3 til 4 år, og de fleste tilfellene oppstår før de er 10 år. En genetisk disposisjon for Wilms svulst hos individer med aniridia er etablert på grunn av sletting av p13 -båndet på kromosom 11.
Historie
Dr. Sidney Farber, grunnlegger av Dana - Farber Cancer Institute, og hans kolleger oppnådde de første remisjonene i Wilms svulst på 1950 -tallet. Ved å bruke antibiotika actinomycin D i tillegg til kirurgi og strålebehandling, økte de kurene fra 40 til 89 prosent.
Bruken av computertomografi for diagnostisering av Wilms svulst begynte tidlig på 1970 -tallet , takket være intuisjonen til Dr. Mario Costici , en italiensk lege. Han oppdaget at i de direkte radiogrammene og i de urografiske bildene kan du identifisere bestemmende elementer for en differensialdiagnose med Wilms svulst. Denne muligheten var en forutsetning for å starte en behandling.
Se også
- Hemihypertrofi
- National Wilms Tumor Study Group (NWTS)
- Perlman syndrom
- Virtuell Karyotype for 1p og 16q LOH
Referanser
Eksterne linker
| Klassifisering | |
|---|---|
| Eksterne ressurser |